200,000+ products from a single source!
sales@angenechem.com


Home > Applications of palladium dibenzylideneacetone as catalyst in the synthesis of five-membered N-heterocycles
Applications of palladium dibenzylideneacetone as catalyst in the synthesis of five-membered N-heterocycles
Navjeet Kaur
Department of Chemistry, Banasthali Vidyapith, Banasthali, Rajasthan, India
Introduction
Heterocycles are very important not only industrially and biologically but also for the functioning of any developed human society.[1,2] The largest classical divisions of organic chemistry are constituted by heterocycles. Many pharmaceutically active compounds and natural products contain heterocycle moieties.[3,4] Transition metal catalyzed reactions are most attractive protocols among a number of new synthetic methodologies because multiply substituted molecules were constructed directly under mild conditions from easily available starting substrates. The development of newer transformations for heterocycle syntheses using atom economical and efficient pathways is a popular research area nowadays.[5–7] One of the most powerful and useful tool in organic synthesis is metal coupling transformation now. Heteroannulation with metal is a convenient and useful method for the synthesis of sulfur containing heterocycles.[8,9]
There is a need for the development of rapid, efficient and versatile strategy for the synthesis of heterocyclic rings. Metal involving methods have gained prominence because traditional conditions have disadvantages such as long reaction times, harsh conditions and limited substrate scopes. In the past decade, metal complexes involving heterocyclic synthesis have become of common use because complicated molecules can be directly built by a metal-assisted reaction under mild conditions from commercial available starting materials.[10–14]
Palladium has a rich organometallic chemistry which has developed over the past 25 years. For products with a high added value, this approach can be cost effective, in that palladium is much cheaper than platinum or rhodium, and high recoveries of palladium can be achieved. The redox behavior of palladium plays an important role in its relevance to organic synthesis, there are at least three other characteristics which enrich the organic chemistry of palladium.
i. The facile rearrangement of trihapto into monohapto ally1 complexes, which creates co-ordinative unsaturation at the metal center and enhances reactivity.
ii. The ability of nucleophile to attack at either metal or ligand sites selectively.
iii. The kinetic lability of palladium-carbon monoxide species which enables carbon monoxide to insert into other palladium-carbon bonds. Among these palladium catalysts, palladium dibenzylideneacetone catalyst is preferred due to following reasons:
I. The reaction proceeds in one step with high regioselective and stereoselective control.
II. The reaction occurs under mild conditions and is not affected by water or air, however if organophosphines are present an inert atmosphere should be used.
III. The reaction is tolerant of most functionalities.[15]
Synthesis of heterocyclic compounds in the presence of transition metal complexes has become common because a metal-catalyzed reaction can form complicated molecules directly from easily available starting materials under mild conditions.[16] In this review, I have focused on the synthesis of several five-membered heterocyclic compounds with nitrogen as the heteroatom in the presence of palladium dibenzylideneacetone catalysts.
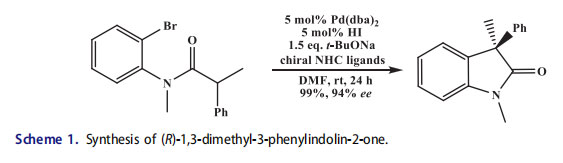
Palladium bis(dibenzylideneacetone)-catalyzed synthesis of five-membered N-heterocycles Kundig et al.[17,18] described the Pd-catalyzed intramolecular a-arylation reactions in the presence of chiral NHC ligands.[19,20] The most successful NHC ligand was examined from this work in Wacker-type oxidative cyclization reactions which afforded the first highly-enantioselective reaction (>90% ee) using a chiral NHC-Pd catalyst (Scheme 1).
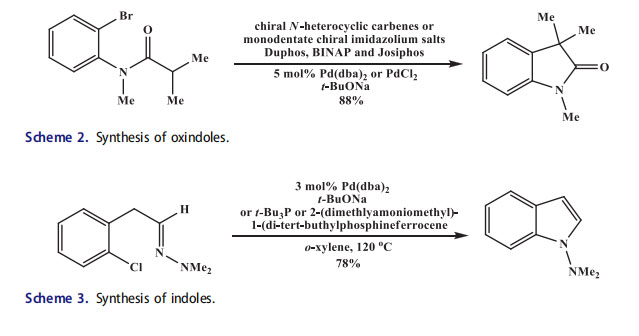
The a-arylation of carbonyl compounds was least investigated transformation, among the several palladium-catalyzed coupling reactions for the formation of C–C bonds, although it was a useful and simple process to synthesize interesting compounds. This simple cross-coupling reaction in the presence of a transition metal catalyst was independently reported by the groups of Hartwig[23] and Buchwald[24] in 1997 which involved a ketone enolate and an aryl halide. Muratake and Natsume[25] at the same time reported the intramolecular a-arylation to synthesize cyclic complex molecules by this short and convenient pathway. Many groups have used chiral N-heterocyclic carbenes in asymmetric intramolecular a-arylation. Hartwig et al.[26] in 2001 first attempted the enantioselective palladium-catalyzed intramolecular reaction of enolates and aromatic bromide in the presence of three different monodentate chiral imidazolium salts. Moderate to good yields of enantioenriched oxindoles were obtained (Scheme 2). With imidazolium salt, the highest selectivity (76% ee) was achieved, which possessed the (þ)-bornyl pattern, the stereogenic center was closer than the (–)-isopinocamphenyl structure. With chiral NHC-ligands the stereoselectivity remained modest and as compared to well-established chiral phosphines like Duphos, BINAP, and Josiphos best result was reported in this field. Glorius et al.[19] in 2002 constructed a C2-symmetric monodentate NHC possessing a two-chiral oxazoline pattern.
Nevertheless, only moderate yields were observed by enantioselective palladiumcatalyzed intramolecular a-arylation. Douthwaite et al.[27] have also performed this reaction with a trans-1,2 diaminocyclohexane spacer/linker containing C2-symmetric diimidazolium salt. However, low selectivity (11% ee) was observed with palladium complex PdCl2.
A variety of nitrogen containing aromatic heterocycles such as benzimidazoles, indoles, quinolines, and pyrrozoles were synthesized by intramolecular C–N bond formation. Watanabe[28] reported an early example for the synthesis of indoles by an intramolecular cyclization of hydrazones with o-chloroarenes (Scheme 3). The reaction occurred from the enamine tautomer through an intramolecular C–N bond formation. This reaction occurred in good yields with bulky electron rich phosphine ligands like t-Bu3P. The enamines and simple imines along with hydrazones also underwent similar cyclizations to synthesize indoles.[29,30] The 1-aminoindoles without substituents in 2 and 3 position were synthesized by cyclization of N,N-dimethylhydrazones of arylacetaldehydes in the presence of phosphane and Pd(dba)2. [31] The chlorine was substituted directly in N,N-dimethylhydrazones of arylacetaldehydes in the presence of an azole, an amine or an arylboronic acid.
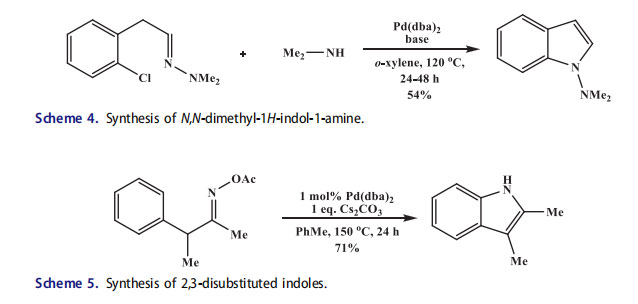
Tetracyclic oxazolocarbazoles (functionalized precursors of carbazoquinocins and antiostatins), hyellazole and carazostatin (polysubstituted carbazole alkaloids), 2-arylindole NK1 receptor antagonists, and vincadifformine and (-)-tabersonine (prominent members of aspirioderma alkaloids) contain indole moiety.[32–35] The isocyanade was transformed into indole by a one-pot synthetic protocol through in situ formed 2-iodoindole[36] (Scheme 4). An intramolecular Stille coupling was also reported for an indole synthesis.[37–44]
Tan and Hartwig[45] reported the amination of arenes with oxime acetates. Under Pd catalysis with Cs2CO3, numerous 2,3-disubstituted indoles were prepared at high temperature and long reaction times (Scheme 5). Moderate yields were obtained for all oxime substrates regardless of sterics and electronics (40–71%). Interestingly, m-OMe substituted oxime was cyclized on the more hindered C–H bond with complete selectivity in high yield (71%). A Pd(II) complex, resulting from N–O oxidative addition, was isolated, characterized and shown to proceed to the desired product upon heating with a base. With this result, the authors suggested that the oxidative addition was the first step, followed by C–H bond insertion and reductive elimination to afford the indole product. Although not mentioned by the authors, the intermediacy of a Pd-nitrene, similar to Buchwald’s proposal for his Cu transformation, should also be viable since the stoichiometric studies cannot rule out this possibility. Following oxidative addition, the Pd species can also form a Pd-nitrene upon elimination of acetic acid; subsequently Pd-nitrene amination afforded the product as well. Soderberg et al.[46,47] reported the synthesis of indoles containing an electron-donating alkoxy group in the 3-position from o-nitrostyrenes through N-heterocyclization (Scheme 6).
Palladium tris(dibenzylideneacetone)-catalyzed synthesis of five-membered N-heterocycles
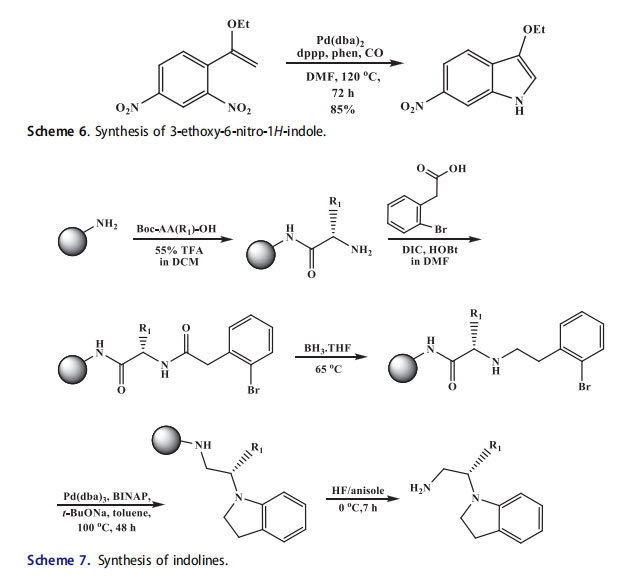
The resin-bound 2-bromophenylacetylated amino acids were used for the solid-phase synthesis of indolines. The secondary amines were formed by reduction of solid-support bound amides with borane. The formed secondary amines by intramolecular cyclization in the presence of palladium catalyst and on cleavage afforded the corresponding disubstituted indulines (Scheme 7).
Di-palladium tris(dibenzylideneacetone)-catalyzed synthesis of five-membered N-heterocycles
Functionalized indoles at C-2 position were synthesized from o-(2,2-dibromovinyl)phenylacetanilide through domino palladium-catalyzed coupling cyclization reaction (Scheme 8).[49] The indole was synthesized through Suzuki coupling which needed the acetanilide derivative (with Boc derivative or free aniline unsatisfactory results were obtained) and Pd2(dba)3 instead of Pd(OAc)2. The o-(2,2-dibromovinyl)-aniline was transformed into a 2-substituted indole by some similarities in a mechanism to that of the last step of the Larock indole synthesis. Different reactivity of the two C–Br bonds in oxidative addition benefited this tandem process.[50,51] The bromovinyl aniline was formed in a first step through Suzuki coupling with substitution of the more reactive trans-bromine atom, then the formed bromovinyl aniline underwent Pd catalyzed intramolecular C–N bond formation. The phosphonylated 2-indolyl derivative was also prepared by this similar method. Moreover, the synthesis of benzofurans was also reported in the same publication using a phenol instead of aniline derivative.[44,52]
The N-fragments were introduced in a single-step cascade sequence onto an acyclic carbon framework for the synthesis of N-functionalized indoles through new palladiumcatalyzed pathway. The o alkynylhaloarenes, (o-haloalkenyl)aryl halides, and gem-dihaloalkenylarenes were developed originally as indole precursors. Willis et al.[53,54] explored the alkenyl triflate substrates and reported that dihalides underwent tandem intermolecular N-alkenylation and intramolecular N-arylation to form indoles. The N-substituted products were afforded in excellent yields in the presence of palladiumbased catalytic system (Scheme 9). Both the E- and the Z-isomers of the starting alkenyl halides were utilized. High yields were also obtained with dichloro substrates. A chloro substituent was introduced at each position of the benzo ring to form the products which were suitable for further synthetic elaboration.[55] Willis et al.[56] used pyridinebased starting compounds for the synthesis of 7-azaindoles. Good yields of products were afforded under similar conditions that were used for the preparation of parent indole. The sterically demanding N-substituents bearing indoles like N-(reverse prenyl)indole were also prepared.[57] The synthesis of natural product demethylasterriquinone A by this approach demonstrated the synthetic utility of this process in which N-(reverse prenyl)indole was used as a key intermediate. The cascade coupling of the vinyl-triflate and aryl-halide bonds in styrene with primary amines formed the indoles.
This reaction occurred with a number of primary amines, carbamates and amides in the presence of a single palladium catalyst (Pd2dba3/Xantphos), despite the fact that two different types of C–X bonds must be activated. It has been found that even vinyl-chlorides participated in this coupling reaction.
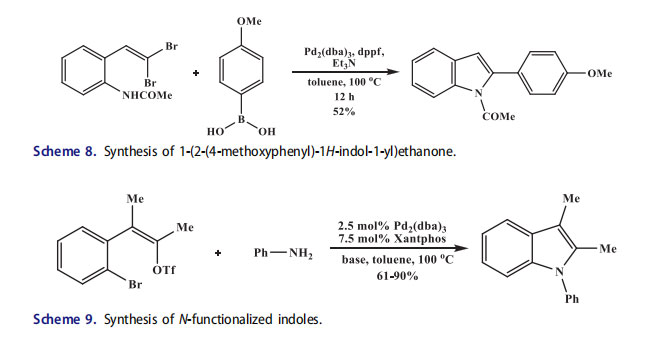
Many reactions have been reported for the preparation of indol derivatives.[59] The anti-Markovnikov hydroamination of o-chloro-substituted 2-alkyl-1-arylalkynes produced arylethylimines by a one-pot synthetic route. The 1,2-disubstituted indoles were synthesized by direct Buchwald-Hartwig coupling of the imine through the formation of tautomeric enamine intermediate under basic conditions (Scheme 10).[60] The substituted benzo[b]furans were accessed through copper-catalyzed O-arylation of ketones formed through hydrolysis of the arylethylimines.[61,62]
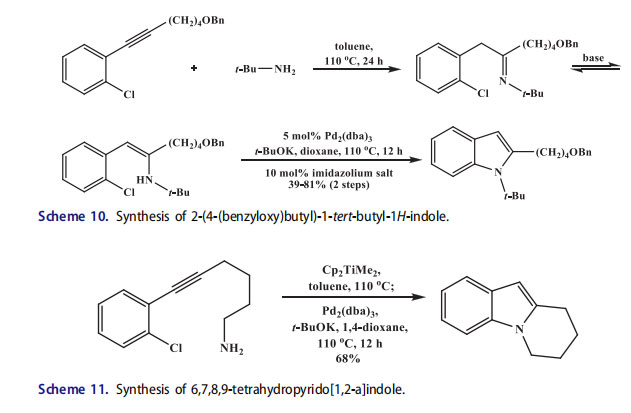
Doye and coworkers[60] reported the one-pot synthesis of N-alkyl and N-arylindoles from 1-(o-chloroaryl)-2-alkyl alkynes which involved the formation of two new C–N bonds (Scheme 11). The alkynes underwent regioselective hydroamination with a primary amine to form imines in the presence of titanium catalyst. The imines were in equilibrium with enamines. Subsequently, the enamines were converted into indoles in good yield by a palladium-catalyzed intramolecular C–N bond forming reaction when the 1,3-bis(2,4,6-trimethylphenyl)imidazolium chloride (precursor of a carbene ligand), Pd2(dba)3, and t-BuOK were added directly to the reaction mixture. The indole was afforded in modest yield (39%) only when the 2-vinyl alkyne was reacted with tertbutylamine, this was due to the worse regioselectivity of the titanium-catalyzed hydroamination of 2-vinyl alkynes in comparison to 2-alkyl alkynes. The scope of this reaction was expanded for the synthesis of cyclic indole derivatives from 1-(o-chloroaryl)-2-(aminoalkyl)alkynes precursors. An intramolecular hydroamination occurred through titanium catalyzed protocol in this case.
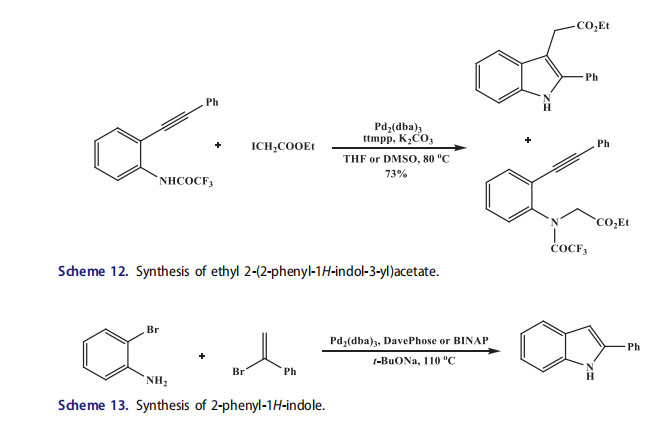
This protocol was extended for the formation of functionalized pyrrole on heteroaromatic scaffolds which offered a new access to pyrroloquinoxaline[63] and azaindole[64] derivatives. In the aminopalladation-reductive elimination alkyl halides such as benzyl bromides and ethyl iodoacetate were also employed for the synthesis of indoles to form the 2-substituted 3-benzylindoles and indolylcarboxylate esters.[65,66] The reaction of heteroaryl, aryl and triflates, or vinyl halides successfully formed the 2,3-disubstituted indoles under the conditions,[67–70] under these same conditions the reaction was unsuccessful and afforded the N-alkyl derivative as main or sole product through a competitive nucleophilic substitution reaction. Noteworthy, the 2-acyl-3-alkylindoles were synthesized from N-alkyl intermediates.[71] The role of bases, ligands, and solvents was investigated to obtain the best results by using Pd2(dba)3, K2CO3, and electron-rich, strongly basic, sterically encumbered ligan tris(2,4,6-trimethoxyphenyl)phosphine (ttmpp)[72] at 80 C in THF. A number of o-alkynyltrifluoroacetanilides were transformed into the desired indole products in satisfactory yields and good selectivity was observed in favor of the palladium-catalyzed cyclization under these conditions (Scheme 12). Similar results were observed with benzyl bromides. The solvent played an important role in this reaction.
For instance, when all the other parameters were kept same and dimethylsulfoxide (DMSO) solvent and o- (phenylethynyl)trifluoroacetanilide were used the indole carboxylate ester was formed in only 12% yield, whereas 64% yield of 2-ethoxycarbonyl-3-benzylindole was reported.[44,49b] Proper conditions were developed for the indolization by the reaction of o-bromoaniline and a-bromostyrene (Scheme 13). The imine was afforded always without indole formation in the presence of BINAP (for the amination of vinyl bromides BINAP is a ligand of choice). When a more active ligand, like bulky electron-rich phosphine DavePhos, was used, the reaction was promoted and indolization occurred with good yield.[52,73]
Itoh and coworkers[74] described the palladium(0)-catalyzed intramolecular cyclotrimerization. Highly substituted dihydroisoindole derivatives were afforded when electron-deficient dialkynes were reacted with DMAD in the presence of Pd2(dba)3 and PPh3 (Scheme 14). Moderate yields of phthalin and isoindoline derivatives were reported by this method. This method was efficiently extended to intramolecular cyclization of triynes to form the tricyclic aromatic systems.[75]
To overcome the nonproductive b-hydride elimination, insertions of alkenes were combined with a fast second reductive elimination step in contrast to carbopalladation sequences of alkynes. Wolfe et al.[76] with the help of this concept have discovered a three component reaction with sequential N-arylation and carbopalladation as elementary steps. The N-arylation produced an alkene-coordinated aryl palladium complex.
The indoline was formed by inserting olefin fragment followed by a reductive elimination (Scheme 15). The nonvolatile, simple, achiral starting material 2-allylaniline was employed. This transformation afforded unsatisfactory results in the presence of dppe or dppb ligands again. However, N-phenyl-2-benzylindoline was formed in 92% yield using Dpe-phos as a ligand.[77] High yields of N-aryl-2 benzylindoline derivatives were also obtained by coupling a wide range of other aryl bromides with 2-allylaniline.[78,79] Author’s prepared two identical aryl groups possessing indolines successfully and it
prompted them to explore the synthesis of two different aryl groups containing related compounds. However, a mixture of mono- and diarylated products was furnished by reacting 2-allylaniline with 1 Eq. of 2-bromonaphthalene. In the presence of (t-Bu)2P(obiphenyl) ligand, high selectivity was observed for monoarylated product (Scheme 16).
The heteroatom possessing bis-allylpalladium analogs were reacted by extending the reaction of bis-allylation. The 2-alkynylisocyanobenzenes, allyl methyl carbonate, and trimethylsilyl azide were reacted by palladium-catalyzed three-component coupling reaction to afford good yields of N-cyanoindoles (Scheme 17).[84] The reaction proceeded through the formation of allylpalladium azide, and subsequently the allylpalladium intermediate was produced by the insertion of divalent carbon of isocyanide into the NPd bond of allylpalladium azide. The allylpalladium intermediate eliminated the nitrogen to form the bis-allylpalladium analog (3-allyl)(3-cyanamido)palladium complex.
Yamamoto et al.[85,86] synthesized 2-substituted 3-allylindoles through cyclization of alkynylbenzenes possessing isocyano and isocyanato moieties in the ortho position. The 2-substituted 3-allyl-N-cyanoindoles were formed when o-alkynylisocyanobenzenes, allyl methyl carbonate, and trimethylsilyl azide were reacted through a three-component reaction in Pd2(dba)3.3CHCl3 and tri(2-furyl)phosphine at 100 C. With a number of substituents in the aryl ring good to allowable yields were reported. The Curtius-like rearrangement of p-allylpalladiumintermediate to the palladium-carbodiimide complex
was a distinctive and interesting aspect of this mechanism. The palladium-carbodiimide and palladium-cyanamide complexes were in equilibrium. The presence of heteroatom possessing bis-p-allylpalladium complex was also suggested. The insertion of alkyne functionality into the Pd-N bond of intermediate followed by a reductive elimination of Pd(0) formed the N-cyanoindole. At 100 C temperature indoles were obtained whereas, reductive elimination of Pd(0) from the palladium-cyanamide complex occurred to produce allyl cyanamides at lower temperature (up to 40 C).[13,44]
Barluenga et al.[87] synthesized indole derivatives by a three-component reaction. This strategy involved a Pd-catalyzed cascade sequence which involved an alkenyl amination, C-arylation and a subsequent intramolecular N-arylation. During the start of the reaction equimolecular amounts of haloalkene, o-dihaloarene, and amines were mixed. The higher reactivity of haloalkene as compared to the haloarene towards the oxidative addition with palladium allowed the unique synthesis of imine intermediate. Further the deprotonation in basic media produced the corresponding aza-allylic anion. The 2-substituted indoles were formed in a subsequent Pd-assisted intermolecular alkylation with the dihalogeno substrate followed by an intramolecular N-arylation. The palladium catalyst was intervened in three different coupling reactions in this cascade reaction. Those three coupling reactions were: intermolecular N-alkenylation, C-arylation, and intramolecular N-arylation (Scheme 18).[88]
Over 100,000 products now available from Angene:
© 2019 Angene International Limited. All rights Reserved.