200,000+ products from a single source!
sales@angenechem.com


Home > Intramolecular Aminoboration of Unfunctionalized Olefins
Intramolecular Aminoboration of Unfunctionalized Olefins
Chun-Hua Yang, Yu-Shi Zhang, Wen-Wen Fan, Gong-Qing Liu, and Yue-Ming Li
Organoboron compounds have found widespread application in a variety of carbon–carbon and carbon–heteroatom coupling reactions. Furthermore, boronic acid moieties have emerged as important functional groups in biologically active compounds. For example, an a-aminoboronic acid is a key structure in the proteasome inhibitor Velcade (bortezomib), and b-aminoboronic acids have been used as a key subunit in peptidomimetics with antitubercular activity.
Traditionally, organoboron compounds have been prepared by transmetalation reactions between organometallic compounds, such as organolithium and organomagnesium reagents, and borates, by the hydroboration of alkenes and alkynes, by the haloboration or aminoboration of alkynes, by the borylation of C=X double bonds, by the borylation of C¢H/C¢X bonds, and by other miscellaneous methods.
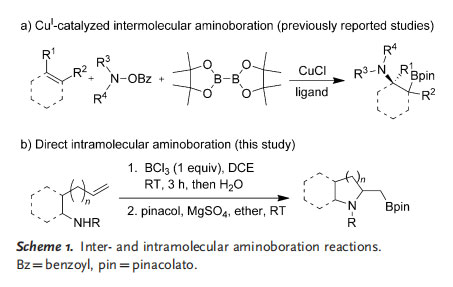
Recently, Hirano, Miura, and co-workers[10] and Tortosa and co-workers[11] described the copper(I)-catalyzed simultaneous addition of nitrogen and boron atoms to C¢C multiple bonds or their equivalents for the preparation of aminoboron compounds. In the presence of a catalytic amount of CuCl, the intermolecular aminoboration of olefins with bis(pinacolato)diboron and O-benzoyl N,N-dialkyl hydroxylamines produced the corresponding b-aminoboron compounds in good to excellent yields (Scheme 1 a). We are interested in developing new, straightforward procedures for the amino-boration of C¢C multiple bonds. Herein, we report our recent results on the direct and catalyst-free intramolecular aminoboration of unfunctionalized C=C double bonds (Scheme 1 b).
In the course of searching for new methods for the amination of C¢C multiple bonds, we found that the intramolecular fluoroamination of unfunctionalized olefins was possible with BF3 as the fluorine source and PhI(OAc)2 as the reaction promoter.[12] We assumed that the direct aminoboration of C=C double bonds should be possible if an N¢B intermediate could be generated during the reaction. In this context, an aminoboration reaction was proposed with BF3·Et2O as the boron source. However, no desired aminoboration product was detected when 1 a was treated with BF3·Et2O. Instead, a very slow intramolecular hydroamination reaction occurred, which was complete after 24 h at 60 8C (Scheme 2).
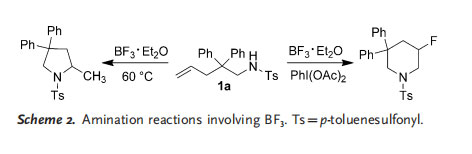
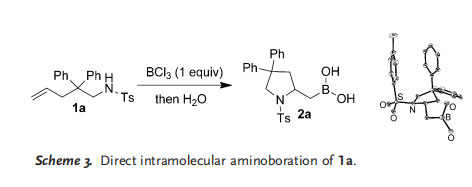
We reasoned that BF3 would interact with the sulfonamide substrate 1 a to form a Lewis pair with either the nitrogen or oxygen atom of the sulfonamido group. The formation of an N¢BF2 bond is difficult owing to the high dissociation energy of B¢F bonds. Instead, Lewis acid mediated reactions took place, and the hydroamination product was isolated. Given that the dissociation energy of a B¢Cl bond is lower than that of a B¢F bond, we reasoned that different reactions should take place if BCl3 was used instead of BF3 under similar reaction conditions. To test this assumption, we added a stoichiometric amount of BCl3 to a solution of 1 a. After 12 h at room temperature, the desired aminoboration product 2 a was obtained (Scheme 3). The structure of 2 a was confirmed by X-ray diffraction.
Encouraged by this result, we optimized the conditions for this direct intramolecular aminoboration reaction. Preliminary results indicated that the reaction medium was crucial for the reaction. Haloalkane solvents were generally suitable, and 1,2-dichloroethane (DCE) gave the most promising result. This solvent was then used as the reaction medium for further studies. Polar aprotic solvents, such as THF, 1,4-
dioxane, DMF, and acetonitrile, were not suitable for the reaction, possibly as a result of their strong interaction with BCl3 and subsequent deactivation of the latter. When the reaction was carried out in toluene, the expected aminoboration product was also obtained. However, the removal of toluene was difficult as compared to the removal of DCE, and reaction in toluene was not pursued further. No significant rate difference was observed when the reaction was carried out at 30 8C instead of 25 8C. Therefore, subsequent reactions were carried out at room temperature without special control of the reaction temperature. A reaction with BBr3 as the boron source gave a similar result. Studies with BBr3 were not continued owing to the relatively difficult conditions needed to handle the reagent.
Having established optimal reaction conditions, we also studied the effect of the protecting group on the course of the reaction. We found that the basicity and nucleophilicity of the nitrogen atom were crucial factors. When the amino group was protected with a tosyl group, the aminoboration product was obtained in good yield. Other protecting groups, such as benzyl, acetyl, trifluoroacetyl, and tert-butoxycarbonyl (Boc) groups, were not suitable, and no aminoboration product was detected when substrates bearing these protecting groups were subjected to the reaction. Deprotection occurred with
the isolation of the corresponding primary amine when a Bocprotected substrate was used. These results indicated that for the aminoboration to proceed, the amino/amido group must have balanced basicity/nucleophilicity. Strongly nucleophilic amino/amido groups will interact more strongly with BCl3, thus leading to the deactivation of both the nitrogen and the boron atom.
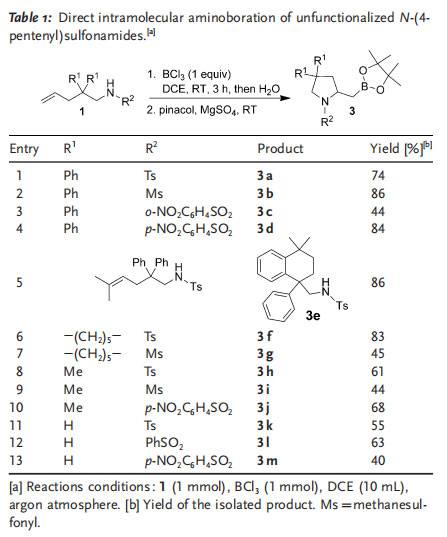
As the purification of boronic acids on a silica-gel column is sometimes problematic, the obtained boronic acids were converted into the corresponding boronates[14] to facilitate product purification. A variety of N-(4-pentenyl)sulfonamide substrates 1 were subjected to the intramolecular aminoboration reaction. After complete consumption of the starting material, the reaction mixture was carefully treated with water, and the crude product was separated from the mixture and treated with pinacol and magnesium sulfate to give the corresponding pinocol boronate. Reactions of p-toluene-, methane-, 2-nitrobenzene-, and 4-nitrobenzenesulfonamide substrates all proceeded readily, and the corresponding boronates were isolated in good overall yields (Table 1).
The reaction was not affected significantly by the Thorpe–Ingold effect (compare the formation of products 3 a, 3 f, 3 h, and 3 k).[15] Substrates with substituents on the main chain, such as 2,2-diphenyl (substrate 1 a), ¢(CH2)5¢ (substrate 1 f), or 2,2-dimethyl (substrate 1 h), were transformed efficiently into the desired aminoboration products, and the aminoboration of substrates without substituents on the main chain (substrates 1 k–m) also gave the corresponding aminoboration products in satisfactory yields (Table 1, entries 11–13).
Substrate 1 e did not undergo the aminoboration reaction. Instead, the Friedel–Crafts alkylation product 3 e was isolated in high yield (Table 1, entry 5).[13] A gram-scale synthesis of 2 a was also carried out to test the scalability of the method: The aminoboration of 1 a on a 5.0 mmol scale afforded boronic acid 2 a in 83% yield. Furthermore, functional-group transformations by oxidation, amination, and Suzuki coupling reactions were possible.
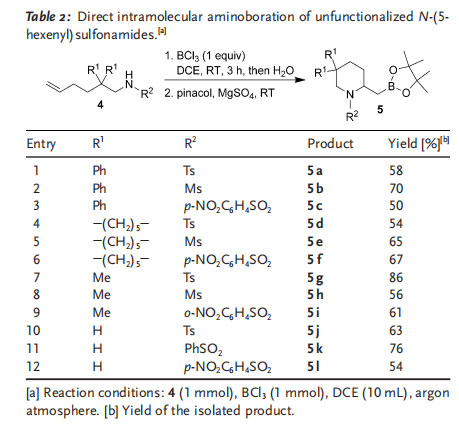
We next turned our attention to the intramolecular aminoboration of N-(5-hexenyl)sulfonamide substrates. Reactions of N-(5-hexenyl)sulfonamide substrates 4 generally proceeded less efficiently than those of N-(4-pentenyl)sulfonamide substrates 1, possibly as a result of the disfavored entropic nature of the cyclization reactions. However, aminoboration products 5 were still isolated in moderate to
good yields under the optimized conditions (Table 2).
Following the synthesis of [(4,4-diphenyl-1-tosylpyrrolidin-2-yl)methyl]boronic acid (2 a), 2-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-ylmethyl)pyrrolidines (3), and 2-(4,4,5,5tetramethyl[1,3,2]dioxaborolan-2-ylmethyl)piperidines (5), we also investigated the synthesis of different 2-(4,4,5,5-tetramethyl[1,3,2]dioxaborolan-2-ylmethyl)-2,3-dihydro-1H-indoles 7 by this method. The desired aminoboration products 7 a–f were isolated in moderate overall yields, and the reaction was not overly sensitive to the electronic effect of substituents on the benzene ring (Table 3).
The formation of the Friedel–Crafts alkylation product 3 e rather than the corresponding aminoboration product when the trisubstituted substrate 1 e was subjected to the reaction (Table 1) indicated the formation of a carbenium cation intermediate. The carbenium cation could be formed by protonation of the substituted C=C double bond with HCl when the corresponding carbenium cation intermediate
showed enough stability. Reactions of terminal alkenes could proceed through the aminoboration pathway since the formation of the corresponding secondary carbenium cation would be slow and less favored.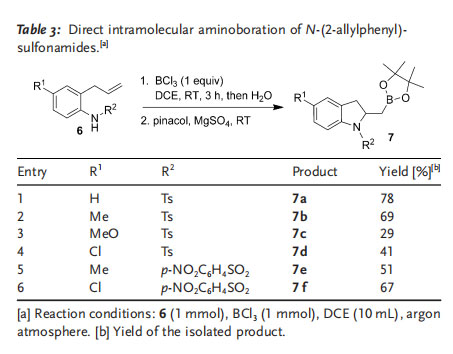
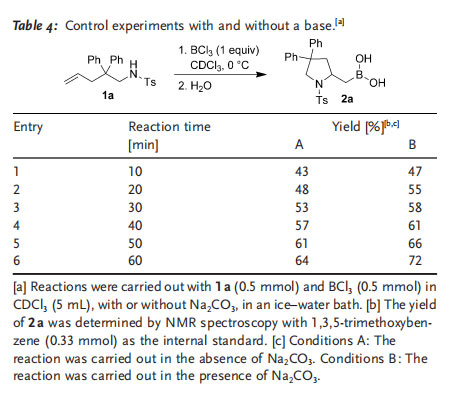
We carried out control experiments to study the possible formation of HCl during the aminoboration reaction. Two reactions were examined under the standard aminoboration conditions, and sodium carbonate was added to one of the two reaction mixtures. The reactions were carried out in an ice–water bath and were sampled every 10 min. The study showed that reactions in the presence of Na2CO3 proceeded slightly faster than reactions without the base (Table 4). This result possibly supports the assumption of the formation of HCl during the reaction. Removal of HCl from the reaction system should help to move the equilibrium to the N¢BCl2 side and speed up the reaction.
NMR spectroscopic experiments were also carried out to study the interaction between BCl3 and the sulfonamide functional group. As a direct NMR spectroscopic study on the interaction between the substrate and BCl3 was difficult owing to the fast conversion of the starting material into the aminoboration product, N-methyl-p-toluenesulfonamide (TsNHMe) was used as a model substrate. 1H NMR spectra showed that the sulfonamide signals changed significantly after the addition of BCl3. Similarly, the 11B NMR signal of BCl3 changed significantly after the addition of TsNHMe. We reasoned that this change was due to the strong interaction between BCl3 and the sulfonamide. DFT calculations were also carried out. Preliminary results indicated that the departure of HCl from the Lewis adduct was the rate-limiting step. However, this step was energetically favored both kinetically and thermodynamically. The intramolecular aminoboration step was found to be a fast step with a low energy
barrier.
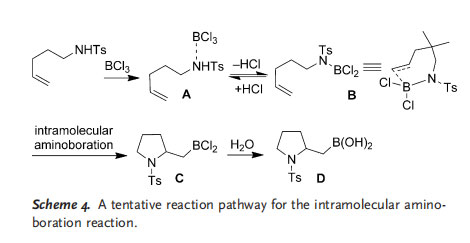
On the basis of previously reported results and these preliminary studies, we propose a possible reaction pathway for the intramolecular aminoboration reaction in Scheme 4: When BCl3 is mixed with the substrate, it interacts with the sulfonamide nitrogen atom to form an LA···NHRTs adduct A. The elimination of HCl from A to give an N¢BCl2 intermediate B is the rate-limiting step. It is also a fast step, and the addition of a base had little effect on the course of the reaction. A downfield shift of the 1H NMR signals for the methylene hydrogen atoms adjacent to the nitrogen atom was observed owing to the attachment of an electron-withdrawing boron group to the nitrogen atom. Intramolecular aminoboration similar to a hydroboration reaction then occurred to give product C, which could be hydrolyzed to afford the final aminoboration product D. We propose that when the substituted substrate 1 e was used, protonation of the C=C bond by the HCl generated in situ led to the formation of a carbenium cation intermediate, which was captured by the phenyl group to yield the Friedel–Crafts alkylation product 3 e.
In summary, we have described the direct intramolecular aminoboration of a variety of N-(4-pentenyl)-, N-(5-hexenyl)-, and N-(2-allylphenyl)sulfonamide substrates by treatment with BCl3. In the presence of BCl3 (1 equiv) as the sole boron source, direct intramolecular aminoboration of these unfunctionalized olefins proceeded readily at room temperature without the use of a catalyst to give the corresponding boronic acids in good yields. The boronic acids could be readily converted into boronates, and the obtained boronates could be further functionalized by oxidation, amination, and Suzuki
coupling reactions. The good yields, mild reaction conditions, and straightforward reaction procedure make this transformation an attractive method for the synthesis of a variety
of useful N-heterocyclic boronic acids and boronates.
Acknowledgements
We acknowledge financial support from the National Natural Science Foundation of China (NSFC 20972072, NSFC 21272121).
Organoboron compounds have found widespread application in a variety of carbon–carbon and carbon–heteroatom coupling reactions:
© 2019 Angene International Limited. All rights Reserved.