200,000+ products from a single source!
sales@angenechem.com


Home > Catalytic asymmetric dearomatization (CADA) reaction-enabled total synthesis of indole-based natural products
Catalytic asymmetric dearomatization (CADA) reaction-enabled total synthesis of indole-based natural products
Chao Zheng* and Shu-Li You *
Covering: 2000 up to 2019.

1. Introduction
The positive interplay between the studies on natural product synthesis and synthetic methodology development constitutes the major theme of fundamental organic chemistry. The fascinating structures of various natural products serve as the inspiration and driving force of endless emerging and evolving unprecedented synthetic methods. On the other hand, natural product synthesis is generally regarded as a premier stage, where a synthetic method can exhibit its practicality and robustness.
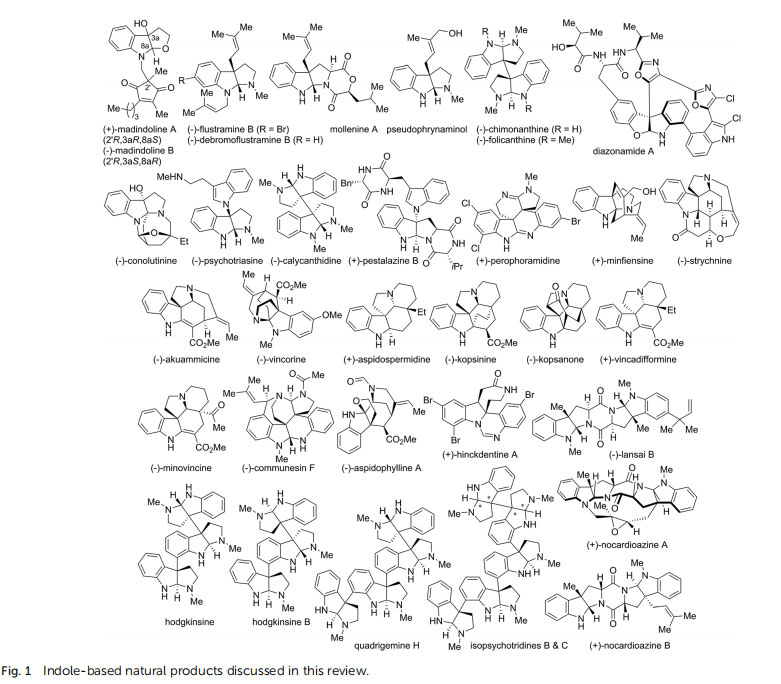
Indole-based polycyclic structures have constantly received intensive research interests due to their wide occurrence in numerous alkaloids that possess diverse biological activities.1–7 Dearomatization of indole derivatives has long been viewed as an attractive strategy to readily access sophisticated spiro or fused polycyclic indoline scaffolds. Accordingly, Qin et al. developed an elegant cascade (or stepwise) reaction of cyclopropanation/ring-opening/iminium (CRI) cyclization of tryptamine derivatives, which enables the efficient assembly of a variety of indoline alkaloids.8 Meanwhile, Ma and co-workers carried out systematic studies on intramolecular dearomative oxidative coupling (IDOC) reactions of indoles and accomplished the total synthesis of many indoline alkaloids.9
In recent years, catalytic asymmetric dearomatization(CADA) reactions have been recognized as powerful methods for the conversion of readily available planar aromatic feedstocks to three-dimensional polycyclic molecules.10–18 Particularly, the synthesis of many indole-based natural products becomes viable with the key ring system and the stereochemistry being established via a CADA reaction. A major advantage of these syntheses is that they do not rely on the chiral pool strategy. The absolute conguration of the product is solely controlled by the chiral catalyst. Therefore, not only natural products themselves, but also their corresponding unnatural enantiomers, diastereoisomers, and even libraries of analogs can be easily synthesized, which is of great importance in medicinal chemistry.
Herein, we summarize the most recent progress on the total synthesis of indole-based natural products enabled by an asymmetric dearomatization reaction as the key step (Fig. 1). Several ring-forming strategies for the construction of key polycyclic scaffolds will be rst highlighted. Then, the various applications of asymmetric dearomatization reactions in the total syntheses of these natural products will be discussed accordingly. Finally, we present a personal perspective of this dynamic research field.
2. General strategies for the formation of key ring systems
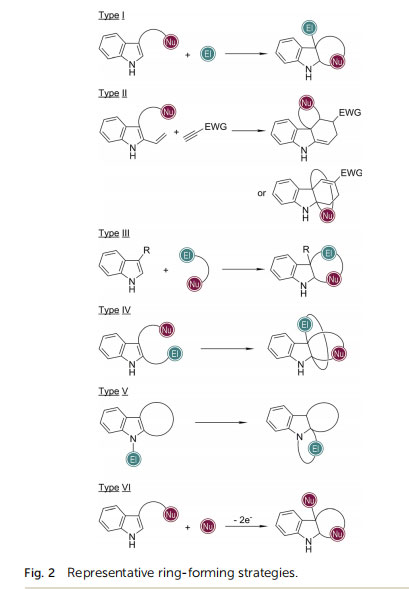
In general, a series of ring-formation strategies have been developed in the total synthesis of indole-based natural products via a CADA reaction (Fig. 2). Most of these strategies take advantage of the inherent strong nucleophilicity of the C3 position of 3-substituted indoles and the electrophilicity of 3,3-disubstituted indolenine cations (types I to IV). By devising appropriately tethered nucleophiles and electrophiles, cascade cyclization reactions may be triggered. On the other hand, the recent enantioselective total synthesis of (+)-hinckdentine A involved a dearomative Hecktype cyclization, which can be regarded as the connection between the C2 position of indole with an electrophile (type V). Notably, the asymmetric dearomatization of indole derivatives via a visiblelight-mediated sequential single-electron transfer process has also emerged, which allowed the formal installation of an external nucleophile at the C3 position of indole with concomitant cyclization (type VI). By employing these strategies, spiro, fused, and even bridged polycyclic scaffolds can be assembled by carefully manipulating the reaction sequence. In the following sections, detailed discussions will be presented on how these strategies
have been applied in the construction of the core structures of indole-based natural products.
3. Type I cyclization
The most frequently employed strategy takes advantage of indole substrates possessing a pendant nucleophile on the C3 position, where tryptamine/tryptophol derivatives are among the commonly used substrates. A Friedel–Cras-type addition with an external electrophile generates a 3,3-disubstituted indolenine intermediate whose iminium moiety is readily trapped by the nucleophilic side chain, yielding the corresponding hexahydropyrrolo[2,3-b]indole (HPI) or tetrahydro-2H-furo[2,3-b]indole core structures, which lays a solid foundation for further synthetic elaborations.
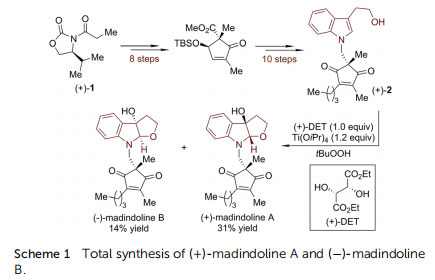
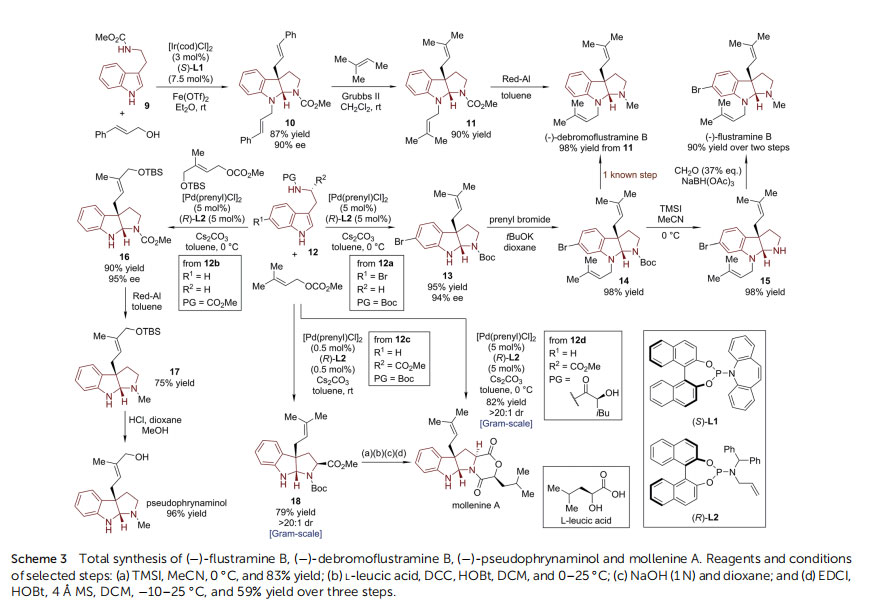
As early as in 2000, Omura, Smith and coworkers furnished the enantioselective synthesis of 3a-hydroxyfuroindoline from tryptophol by employing the Sharpless asymmetric epoxidation protocol (Scheme 1). This strategy was successfully applied in their study towards the total synthesis and the absolute conguration assignment of (+)-madindoline A and (+)-madindoline B, which are potent inhibitors of interleukin 6 (IL-6).19 N-Alkylated tryptophol (+)-2 was prepared from oxazolidinone (+)-1 via 18 linear steps. The (+)-DET/Ti(OiPr)4-promoted Sharpless asymmetric epoxidation/cyclization, a process involving the asymmetric dearomatization of indole, served as the nal step to complete the synthesis of (+)-madindoline A (31% yield) and its diastereoisomer ()-madindoline B (14% yield). ()-Flustramine B and ()-debromoustramine B belong to a family of marine alkaloids isolated from the Bryozoa Flustra foliacea, which possess skeletal and smooth muscle relaxant activity and signicant butyrylcholinesterase inhibitory activity, respectively.20 Their embedded HPI core structure makes them popular targets in recent asymmetric total synthesis studies.21–23
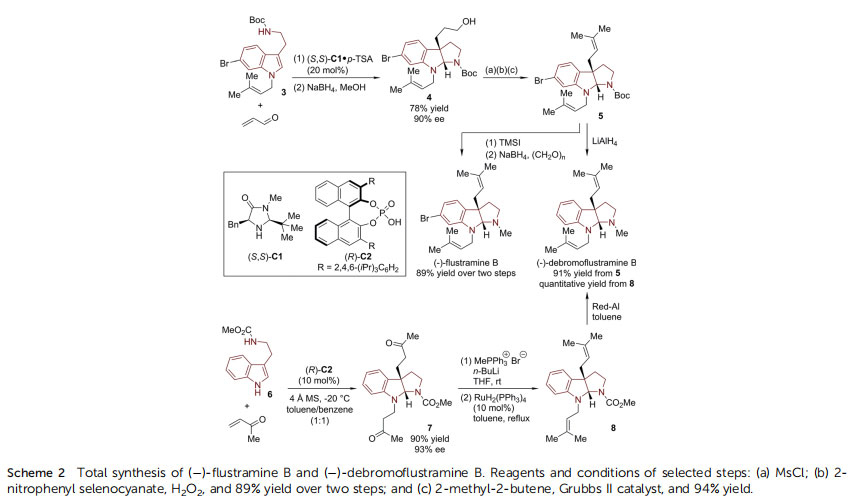
In 2004, the MacMillan group reported the enantioselective synthesis of ()-ustramine B and ()-debromoustramine B(Scheme 2).24 In the presence of an imidazolidinone catalyst(S,S)-C1,
25 6-bromo-substituted tryptamine derivative 3 reacted smoothly with acrolein in a cascade dearomative addition/cyclization sequence. HPI compound 4 with a pendant primary alcohol group was achieved in 78% yield with 90% ee aer in situ reduction by NaBH4. The 3-hydroxy propyl sidechain was readily converted to a prenyl moiety in a three-step, two-pot sequence. The primary alcohol in 4 was rst mesylated and then transformed into terminal alkene by treatment with 2-nitrophenyl selenocyanate/H2O2. A crossmetathesis with 2-methyl-2-butene by Grubbs II catalyst incorporated the prenyl moiety in 5. Finally, ()-ustramine B and()-debromoustramine B were obtained via selective reduction of the N-Boc group and removal of the 6-bromo substituent in the latter case.
In 2012, Zhang and Antilla reported the asymmetric synthesis of ()-debromoustramine B from tryptamine derivative 6 and methyl vinyl ketone (MVK) using a chiral phosphoric acid (CPA) catalyst (Scheme 2).26 With TRIP-CPA (R)-C2 (ref. 27) as the optimal catalyst, the HPI core of 7 was assembled with two MVK segments incorporated at the N1 and C3a positions in good yield (90%) and enantioselectivity (93% ee). Subsequently, the two methyl ketone moieties were transformed into two prenyl groups of 8 via sequential Wittig reaction/olen isomerization. ()-Debromoustramine B was obtained in quantitative yield via Red-Al reduction of 8. During their continuous efforts towards transition-metalcatalyzed asymmetric allylic dearomatization reactions,12 You's group developed a series of efficient asymmetric syntheses of the ustramine family of natural products (Scheme 3).28 With [Ir(cod)Cl]2 as the Ir-precursor, Carreira P/olen ligand L1 (ref. 29) as the optimal chiral ligand, and Fe(OTf)2 as the promoter, the dearomatization reaction of tryptamine derivative 9 occurred with cinnamyl alcohol, leading to N1,C3a-biscinnamyl substituted HPI product 10 in good yield (87%) with high enantioselectivity (90%ee). The cinnamyl groups could be converted to prenyl groups through cross-metathesis reaction by the Grubbs II catalyst. The nal reduction of the N-CO2Me group furnished the total synthesis of () debromoustramine B. Very recently, You and coworkers discovered an enabling Pd-catalyzed enantioselective dearomative prenylation of indole derivatives.30 By employing a Pd-complex derived from [Pd(prenyl)Cl]2 and a newly synthesized chiral phosphoramidite ligand allylphos (L2), prenyl carbonate could react with a large array of indole derivatives 12.
In particular, the reaction of the 6-bromo-substituted tryptamine derivative 12a afforded the prenylated HPI product 13 in excellent yield (95%) and enantioselectivity (94% ee). The following Nprenylation reaction resulted in the formation of compound 14, which is a known precursor of ()-debromoustramine B.24 Meanwhile, ()-ustramine B could also be prepared from 14 aer deprotection of the N-Boc group and reductive amination. On the other hand, the reaction of 12b with a different prenyl precursor bearing a tethered silyl ether group provided the dearomatized product 16. The following reduction of the ester group and removal of the silyl group accomplished the synthesis of ()-pseudophrynaminol. In addition, with the readily available tryptophan derivative Boc-L-Trp-OMe 12c as the starting material, the synthesis of prenylated HPI product 18 could be realized on the gram-scale with a catalyst loading as low as 0.5 mol%. Subsequent protecting group manipulation and coupling with Lleucic acid furnished the total synthesis of mollenine A, which corrected the assignment of its absolute conguration.31–33
Notably, this Pd-catalyzed enantioselective dearomative prenylation reaction tolerates highly functionalized indole derivatives as substrates. Under the standard conditions, the L-Trp-Lleucic acid 12d was involved in a cascade dearomative prenylation/cyclization/lactonization reaction, which allowed the one-step synthesis of mollenine A on the gram-scale, and also a library of analogues of mollenine A to be built from the corresponding tryptophan derivatives synthesized by reacting with various a-amino acids or chiral a-hydroxyl carboxylic acids. ()-Diazonamide A belongs to a structurally unique class of
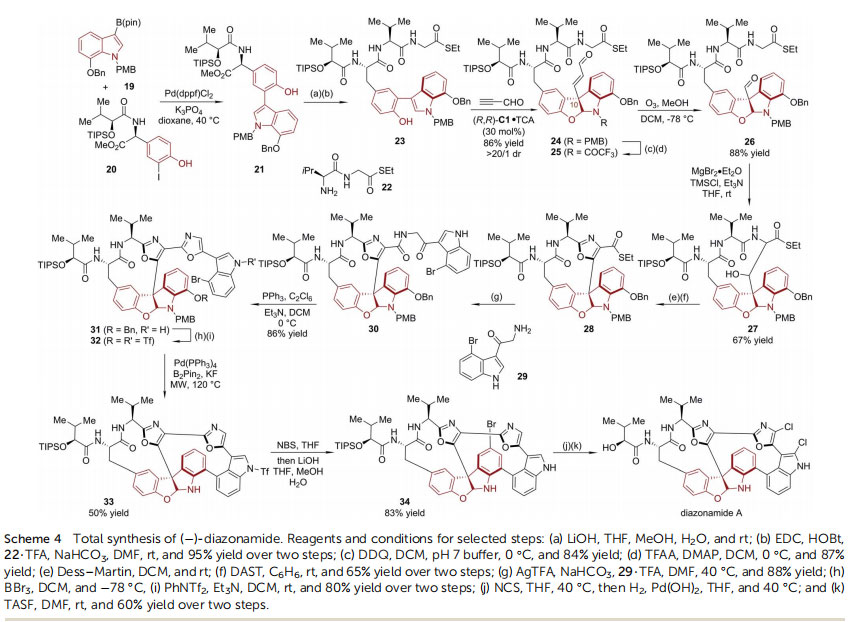
secondary metabolites isolated from the colonial marine ascidian Diazona angulata and is a potent antimitotic agent.34,35
In 2011, MacMillan and coworkers reported the asymmetric total synthesis of diazonamide A (Scheme 4).36 Starting from functionalized indole boron ester 19 and 2-iodophenol 20, Suzuki coupling furnished 2-(indol-3-yl)phenol 21, whose complexity was further enriched to 23 in two steps. With 23 and propynal as substrates, the imidazolidinone catalyst (R,R)-C1-enabled asymmetric dearomative addition/cyclization strategy was applied to assemble the key tetracyclic core with concomitant establishment of the quaternary chiral center at the C10 position, leading to the key intermediate 24 in good yield (86%) with excellent diastereoselectivity (>20 : 1 dr). Aer the exchange of the N-protecting group from PMB to TFA, ozonolysis furnished aldehyde 26. Subsequently, an intramolecular aldol reaction closed the 13-membered ring, affording 27 as a single diastereoisomer. The central oxazole ring (28) was then formed via a two-step oxidation/cyclodehydration sequence. In the presence of AgTFA, the thioester functionality of 28 was displaced by amine 29. The resulting intermediate 30 underwent a second cyclodehydration, furnishing bisoxazole 31. Aer debenzylation and treatment with PhNTf2, the bromo-bistriate intermediate 32 was obtained. The tandem Pd-catalyzed borylation/annulation of 32 occurred smoothly with B2pin2/KF under microwave heating, leading to the full installation of the
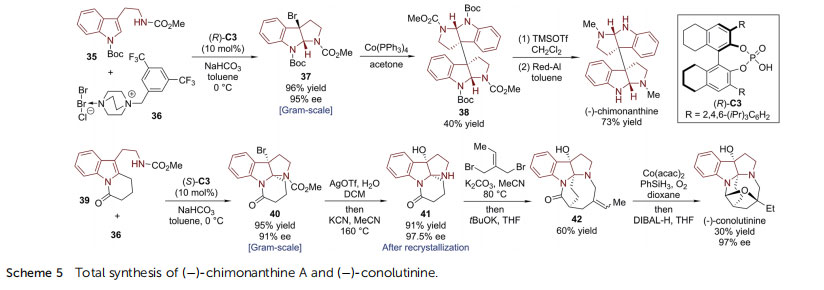
carbon framework of ()-diazonamide A. The nal selective chlorination was achieved in two steps, employing Br as a transient protecting group for the C5 position of the activated indoline ring. ()-Chimonanthine is a dimer of HPI units connected by a C3–C30 bond, which has long been known as a synthetic target.37,38 In 2013, Xie, Lai, Ma and coworkers reported the concise enantioselective total synthesis of ()-chimonanthine based on the asymmetric dearomative bromocyclization of tryptamine 35 (Scheme 5).39 By utilizing DABCO-derived bromine salt 36 as the electrophilic bromination reagent and chiral phosphoric acid 8H-TRIP (R)-C3 as the catalyst, the C3-brominated HPI compound 37 was delivered on the gramscale in excellent yield (96%) with high enantioselectivity(95% ee).40 Next, the Co-catalyzed homodimerization of 37 afforded 38 in 40% yield. The latter possesses the entire skeleton of ()-chimonanthine with correct stereochemistry.41,42
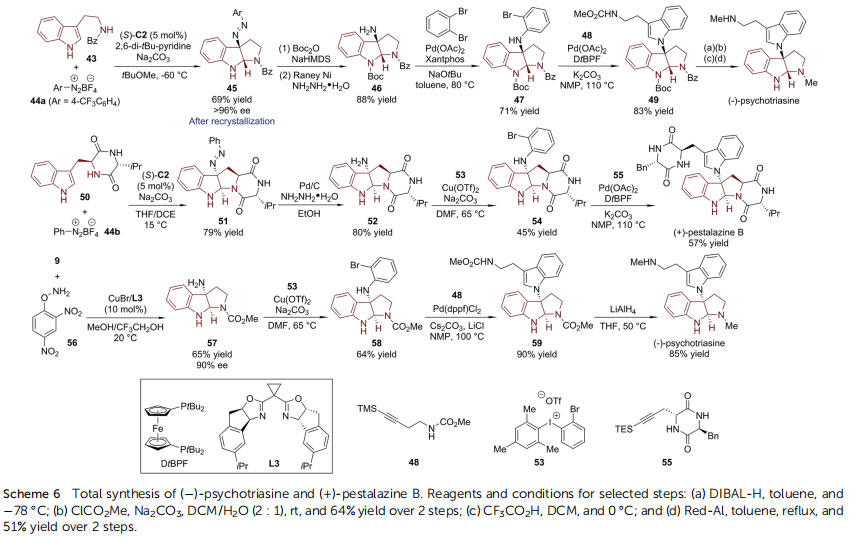
Simple removal of the protecting groups from 38 led to the target molecule. Subsequently, Lai, Xie and coworkers realized the asymmetric total synthesis of ()-conolutinine, a terpenoid indole alkaloid isolated from Malaysian Tabernaemontana43 with a similar asymmetric dearomative bromocyclization reaction (39 / 40) as the key step (Scheme 5).44 Hydrolysis of the bromide and removal of the N-CO2Me protecting group of 40 provided 41. The enantiopurity of 41 was improved to 97.5% ee aer recrystallization. The desired eight-membered ring was constructed via double SN2 reactions of 41 with 1-bromo-2-(bromomethyl)but-2-ene. The nal ring in ()-conolutinine was closed aer Co-mediated hydration of the exocyclic double bond followed by reduction of the amide by DIBAL-H. 3a-Amino-pyrroloindoline is another type of important core structure widely found in indole alkaloids and pharmacological compounds. In 2015, Deng, Liao and coworkers developed the asymmetric synthesis of 3a-amino-pyrroloindolines from the corresponding tryptamine derivatives and aryl diazonium salts catalyzed by a chiral phosphoric anion-paired catalyst(Scheme 6).45 With (S)-C2 as the chiral catalyst and Na2CO3 and 2,6-di-tBu-pyridine as the bases, the reaction between 43 and 44a produced C3-diazenated pyrroloindoline 45 in high enantiopurity (>96% ee).46 The N]N double bond could be cleaved
with hydrazine hydrate in the presence of RANEY® Ni to afford the 3a-amino-pyrroloindoline compound 46, which underwent Buchwald–Hartwig amination with 1,2-dibromobenzene in the presence of a Pd-catalyst derived from Xantphos, leading to 47. Next, Pd-catalyzed cyclization of 47 with alkyne 48 afforded tryptamine dimer 49, from which routine protecting group manipulations delivered ()-psychotriasine.47 Under slightly modied conditions, the cyclic dipeptide 50 could couple with the phenyl diazonium salt 44b to give the dearomatized product 51 in 79% yield. Subsequently, the diazo group was reduced to primary amine 52 by hydrazine hydrate and Pd/C. Aer a series of screenings, the ortho-brominated hypervalent iodine reagent 53 was identied as a suitable coupling partner for the Cucatalyzed C–N bond formation, leading to 54 in reasonable yield (45%). The total synthesis of (+)-pestalazine B48,49 was completed aer nal cyclization of 54 with alkyne 55 (Scheme6). Shortly aer, Tang, You and coworkers reported the Cucatalyzed asymmetric dearomative amination reaction of tryptamines (Scheme 6).50 In the presence of a Cu-catalyst ligated by the novel chiral BOX ligand L3, 3a-amino-pyrroloindoline 57 could be directly obtained in a reasonable yield (65%) and good enantioselectivity (90% ee) from tryptamine derivative 9 with O-(2,4-dinitrophenyl)hydroxylamine (DPH) 56 as the amination reagent. In this regard, ()-psychotriasine was readily prepared from 57 through a known C–N bond formation/Larock cyclization/reduction sequence.
In 2013, Zhu and MacMillan disclosed that the reaction between tryptamides and diaryliodonium salts by a chiral Cu–BOX complex could lead to the facile synthesis of aryl pyrroloindolines through an arylation/cyclization strategy.51–53 This method paved the way for their later discovery of the collective synthesis of higher-order polypyrroloindoline natural products(Scheme 7).54 The key protocol involved in these syntheses is the iterative homologation of the pyrroloindoline segment by a twostep reaction, which connects the two neighboring pyrroloindolines between their C3 and C70 positions.
For example, in the presence of a chiral Cu-catalyst (S,S)-C4, the reaction of the nucleophilic n-order oligomer 60 with the electrophilic aryl indolyliodonium salt 61 led to compound 62 with the formation
of a stereochemically dened C3–C70 bond. Subsequently, the carbonyl group of the a-ketoamide side chain of 62 was reduced by Hantzsch ester, re-establishing a nucleophilic tryptamide moiety in the (n + 1)-order oligomer 63 for the next-round iteration. The synthetic sequence was terminated via a final head-to-head tryptamine dimerization with N1-Bn-N-Moctryptamine facilitated by N chlorophthalimide, which installed the desired pseudo-meso “cap” through the formation of the requisite C3–C30 bond (64). With this synthetic plan, the MacMillan group furnished the total synthesis of hodgkinsine (>450mg), hodgkinsine B,55–57 idiospermuline,58 quadrigemine H59 and isopsychotridines B and C.60,61 Since the stereochemistry of the reaction sequence was controlled by the chiral catalyst, this method was also capable of preparing two putatively unnatural quadrigemine analogs.
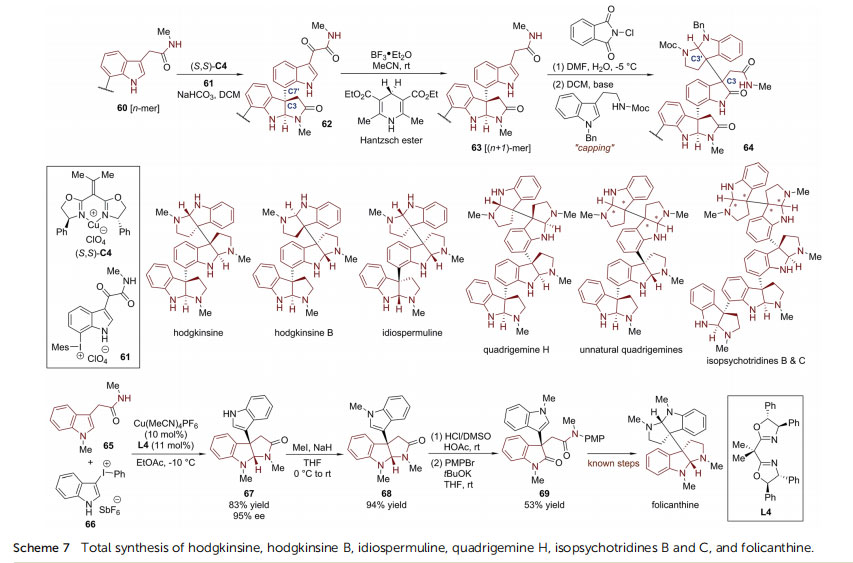
Notably, prior to MacMillan's report on the collective synthesis of polypyrroloindoline natural products,54 You and coworkers developed Cu-catalyzed asymmetric dearomatization reactions of tryptamides with 3-indolylphenyliodonium salts (Scheme 7).62 With the Cu-complex derived from the chiral BOX ligand L4 as the optimal catalyst, the reaction between tryptamide 65 and 3-indolylphenyliodonium salt 66 provided straightforward access to 3-(3a-indolyl)-hexahydropyrroloindoline 67 in high yield (83%) with excellent enantioselectivity (95% ee). Aer routine protecting group manipulations and hydrolysis of the aminal moiety, 67 was readily converted to chiral oxindole 69, a known precursor to folicanthine.63
4. Type II cyclization
The MacMillan group developed a Diels–Alder/aminative cyclization strategy capable of constructing polycyclic indoline skeletons. In the presence of a chiral imidazolidinone catalyst, 2-vinyl tryptamine derivatives work as a diene in an enantioselective Diels–Alder reaction with an unsaturated aldehyde. Then the nucleophilic attack of the pendant amine nucleophile to an appropriate electrophilic site closes the fused or bridged cyclic skeleton. In their nine-step enantioselective synthesis of (+)-min-ensine, a structurally unique Strychnos alkaloid,64 MacMillan and coworkers applied the Diels–Alder/aminative cyclization strategy to assemble the tetracyclic carbazole framework of this natural product (Scheme 8).65 2-Vinylsulde-substituted tryptamine 70 was allowed to react with propynal in the presence of
a catalytic amount of (R,R)-C5. The cyclohexadienyl intermediate I isomerized into the indolenine intermediate II, which was readily trapped by the amine side chain via a 5-exo cyclization, forging the bridged tetracyclic pyrroloindoline 71 in high yield (87%) with excellent enantioselectivity (96% ee).
Treatment of 71 with TESOTf removed the N-Boc protecting group and the secondary amine formed in 72 underwent reductive amination with 4-(tert-butylthio)but-2-ynal 73, delivering propargylic sulde 74. In this design, the incorporation of a 1-methyl sulde group in 70 not only accelerated the Diels–Alder reaction, but also served as a latent handle for the subsequent radical cyclization facilitated by AIBN/tBu3SnH, which is required in the formation of the nal ring (74 / 75). (+)-Minensine can be obtained from 75 via simple manipulations in two steps.
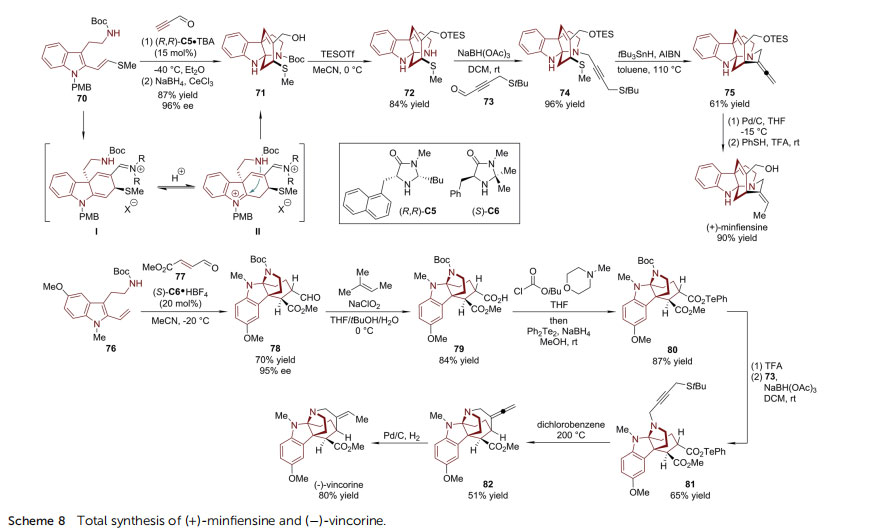
Subsequently, Horning and MacMillan reported the highly efficient enantioselective synthesis of ()-vincorine, the parent compound of an akuammiline alkaloid family,66 by applying the same strategy (Scheme 8).67 With a different imidazolidinone catalyst (S)-C6, the sequential Diels–Alder/aminative cyclization reaction of 2-vinyl-substituted tryptamine 76 with the functionalized a,b-unsaturated aldehyde 77 led to the key intermediate 78 in 70% yield with 95% ee. The aldehyde group in 78 was oxidized to acid 79 and then transformed into acryl telluride 80.
As employed in the synthesis of (+)-minensine,65 a propargylic sulde moiety was introduced in 81 via reductive amination. The construction of the challenging seven-membered azepanyl ring in 82 was realized based on an intramolecular radical cyclization between the propargylic sulde and the acyl telluride moieties of 81 under thermal conditions. The nal hydrogenation reaction of the allene group of 82 completed the synthesis of ()-vincorine.
The power of this Diels–Alder/aminative cyclization strategy was further exemplied with the collective synthesis of Strychnos, Aspidosperma and Kopsia alkaloids reported by the MacMillan group (Scheme 9).68 The key step in this synthetic sequence is the generation of tetracyclic spiroindoline 84, common advanced intermediates of the target natural products.
The syntheses of 84 relied on the coupling of 2-vinylselenidesubstituted tryptamines 83 with propynal by the chiral imidazolidinone catalyst C5. The methylselenide group of III had a high propensity to undergo b-elimination, which afforded the highly conjugated iminium IV. Thus, the desired advanced intermediates 84 could be generated in good yields (82–83%) with high enantiopurity (97% ee) aer the facile Michael addition of pendant amine in IV. The synthesis of ()-strychnine, the best-known member of the Strychnos family,69 started from 85, which could be obtained from (3aR,11bR)-84a via three
steps. The reduction of the ester group of 85 and the introduction of a vinyl iodide moiety led to 87, from which a Heck cyclization/lactol formation/N-deprotection afforded the Wieland–Gumlich aldehyde 89, a well-known precursor of ()-strychnine.70 Notably, the N-PMB group was crucial for the efficiency of these reaction sequences. Finally, enantioenriched()-strychnine was delivered when 89 was heated in a mixture of malonic acid, acetic anhydride and sodium acetate.
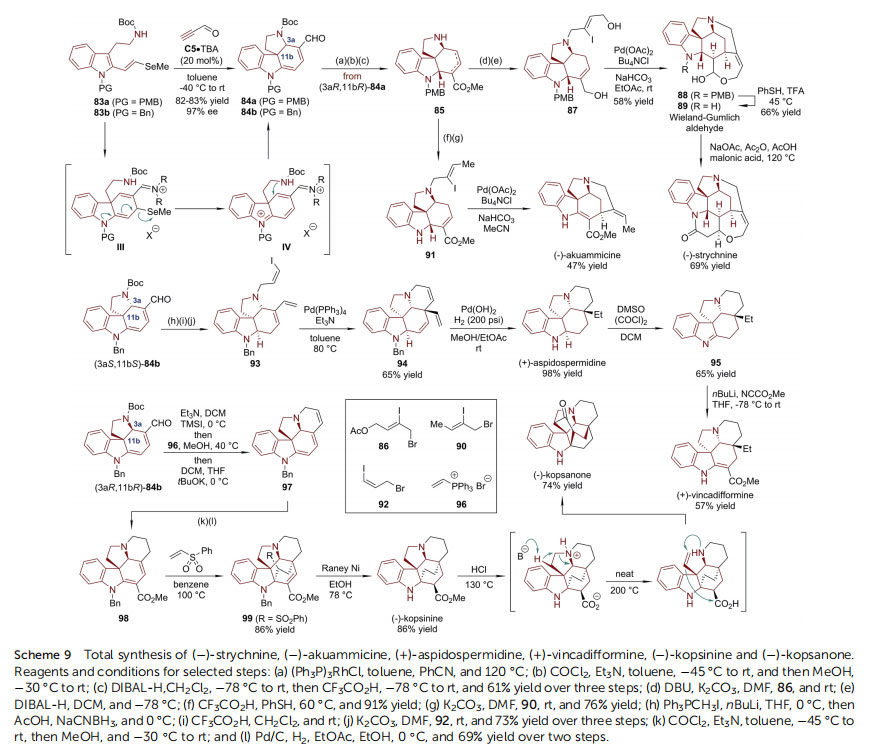
In addition, the synthesis of ()-akuammicine71 could also be readily accomplished from 85. Aer the removal of the NPMB group and isomerization of the C]C double bond into conjugation with the ester moiety by TFA/thiophenol, Nallylation incorporated a vinyl iodide group in 91, which is a direct precursor of ()-akuammicine via Heck cyclization. Notably, by employing the slight variant (3aS,11bS)-84b as an entry point, only simple functional group manipulations were required to realize the synthesis of (+)-aspidospermidine72 and(+)-vincadifformine,73 which are among the most highly sought Aspidosperma alkaloid targets. Starting from (3aS,11bS)-84b, Wittig reaction of the dienal moiety followed by deprotection of the N-Boc group and N-allylation reaction led to 93. The regioselective Heck cyclization of vinyl iodide with the trisubstituted endocyclic C]C double bond furnished 94. With Pd(OH)2 on carbon as the catalyst, global hydrogenation and debenzylation were achieved simultaneously, completing the total synthesis of(+)-aspidospermidine. In addition, the Swern oxidation of(+)-aspidospermidine afforded imine 95, which was readily transformed into (+)-vincadifformine when treated with nBuLi followed by methyl cyanoformate. Moreover, the concise syntheses of the highly caged Kopsia alkaloids ()-kopsinine74 and ()-kopsanone75 were made possible based on (3aR,11bR)-84b. The removal of the N-Boc group triggered a conjugate addition to vinyl triphenylphosphonium bromide 96 and subsequent Wittig olenation, leading to cyclic triene 97. It was reacted with phosgene/ methanol, accomplishing enamine a-carbomethoxylation.
Selective hydrogenation by Pd/C delivered diene 98, which underwent Diels–Alder reaction with phenylvinyl sulfone, furnishing 99. Desulfonylation, hydrogenolysis of the N-Bn group and diastereoselective alkene reduction were realized in one step with RANEY® Ni to yield ()-kopsinine. It was found that this molecule could be transformed to ()-kopsanone efficiently via an acid-promoted skeleton rearrangement. It should be emphasized that the Diels–Alder/aminative cyclization strategy enabled the enantioselective total synthesis of six structurally diverse natural products in only 9 to 12 steps with
unprecedented high efficiency.
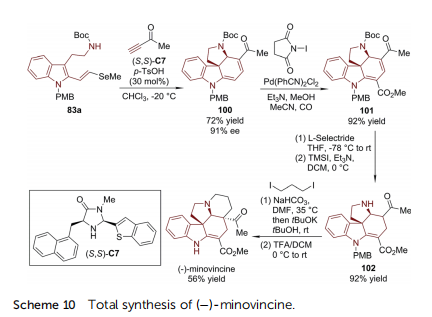
The dienophile of the Diels–Alder/amine cyclization strategy was further extended to propargylic ketone (Scheme 10).76 The reaction between 2-vinylselenide-substituted tryptamine 83a and 3-butyn-2-one was facilitated by chiral imidazolidinone catalyst (S,S)-C7, leading to tetracyclic spiroindoline 100 bearing an exocyclic ketone substituent (72% yield, 91% ee). Concise downstream elaborations resulted in the total synthesis of()-minovincine.77 The Pd-catalyzed carbomethoxylation of 100 was realized in the presence of CO/MeOH to afford 101. 1,4-Conjugated reduction of 101 was achieved with bulky Lselectride followed by deprotection of the N-Boc group, which gave rise to 102. The synthesis of ()-minovincine was completed aer the closing of the nal ring with 1,3-diiodopropane and debenzylation from the indoline nitrogen atom.
5. Type III cyclization
The type III cyclization strategy employs the reaction of C3-substituted indoles with an external amphiphilic reagent in a concerted manner or in separate steps. The connections between the C3-position of the indole ring to the electrophilic site of the amphiphilic reagent and the C2 position to the nucleophilic site furnish the dearomatization process with the fast construction of molecular complexity.
The most signicant example comes from the synthesis of ()-lansai B78 and (+)-nocardioazines A and B,79,80 bis(-pyrroindolines) joined through a central diketopiperazine(DKP) ring, which was reported by Wang and Reisman(Scheme 11).81 In 2013, Reisman's group developed an asymmetric dearomative [3 + 2] annulation reaction between C3-substituted indoles and 2-amidoacrylates 103 using a BINOLderived chiral Sn-complex.82 The synthesis of these natural products is quite convergent. Starting from N-methyl skatoles 104 and 107, the Sn-complex-catalyzed asymmetric dearomative[3 + 2] annulation reaction with 103a led to pyrroloindoline compounds in high yields with excellent diastereo- and enantioselectivities (85% yield, 92% ee, and 14 : 1 dr for 105 and 79% yield, 93% ee, and 12 : 1 dr for 108) respectively. The utilization of 2,6-dibromophenol is believed to be benecial for the turnover of the catalyst, while not being acidic enough to protonate the enolate intermediate in a racemic fashion. Subsequent twostep protecting-group manipulations afforded naked amino acids 106 and 109 as the nal coupling partners, respectively.()-Lansai B was afforded (38% yield) by the assembly of the central DKP ring through a double amide condensation reaction, with the concomitant formation of homodimers of 106 and 109 (about 20% yield for each homodimer).
The synthetic route towards (+)-nocardioazine B is rather similar to that of ()-lansai B (Scheme 11). With different substituted indoles (110 and 113) as starting materials, and slightly varied conditions in the steps for asymmetric dearomative [3 + 2] annulation reactions and following protecting group manipulations, orthogonally protected amino acids 112 and 115 were obtained smoothly, respectively, which participated a stepwise cyclization, assembling the central DKP ring in 116. The nal Pd-catalyzed deallylation and incorporation of a prenyl moiety via cross-metathesis furnished the total synthesis of (+)-nocardioazine B.
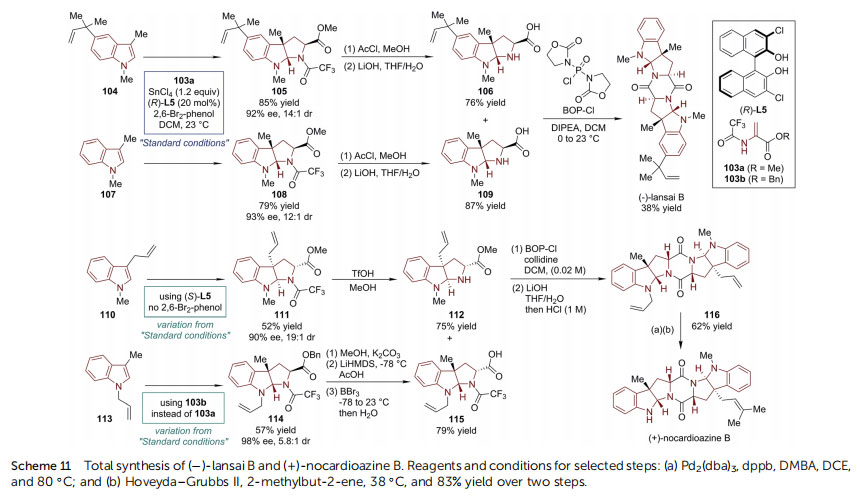
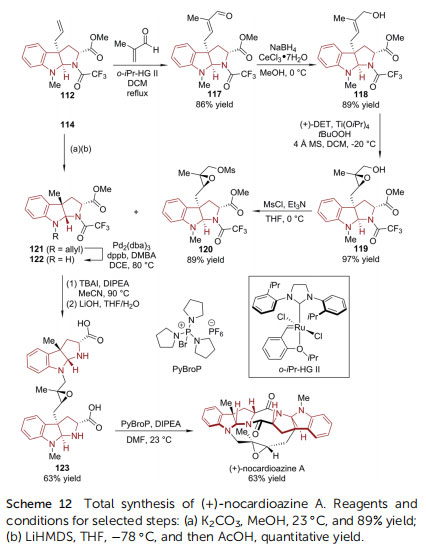
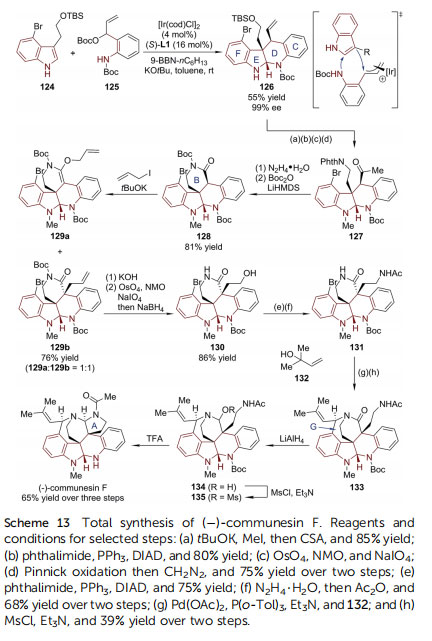
Notably, (+)-nocardioazine A has a more complicated caged structure, in which the two HPI units are connected by a trisubstituted epoxy moiety. The strategy of forging the epoxy moiety in the nal stage was not successful, only leading to an epimer of the target molecule. In this regard, another synthetic plan involving early epoxidation was executed (Scheme 12). Cross-metathesis of the previously synthesized HPI derivative 112 with a-methyl acrolein afforded enal 117. Subsequent reduction, epoxidation, and mesylation provided intermediate 120. Simultaneously, the other coupling partner 122 was prepared from 114 via a three-step sequence (transesterication/epimerization/deallylation). An SN2 reaction mediated by TBAI and Hunig's base connected ¨ 120 and 122. The following saponication of methyl ester afforded bis(amino acid) 123. A nal macrocyclization through double amide condensation furnished (+)-nocardioazine A with high efficiency.()-Communesin F belongs to a family of complex polycyclic indole alkaloids isolated from marine and terrestrial Penicillium fungi.83 Its signicant cytotoxicity and insecticidal activity, together with its intriguing structural complexity (heptacyclic core, two aminal groups, and ve stereogenic centers including two vicinal quaternary ones) make this molecule a popular synthetic target.84,85 Recently, Yang and coworkers reported the rst catalytic asymmetric synthesis of ()-communesin F (Scheme 13).86 During the early stage of the synthesis, the N,Naminal-containing CDEF tetracyclic skeleton (126) was constructed via an asymmetric allylic substitution/cyclization reaction between silyl-protected tryptophol 124 and anilinederived allylic electrophile 125, enabled by the Carreira catalyst consisting of [Ir(cod)Cl]2 and chiral P/olen ligand (S)-L1. It was identied that the judicious choice of the activator for the indole substrate was crucial for the reaction efficiency. The utilization of 9-BBN-nC6H13 in combination with tBuOK not only led to the highest enantioselectivity of the reaction (99%ee), but also caused the reaction to proceed smoothly at a 5 mmol scale. In the following four steps, the silyl ether was converted to imide and the terminal olen was oxidized to ester (127). Upon treatment with N2H4$H2O, the spontaneous lactamization closed the B ring in 128. When reacted with allyl iodide, 128 was transformed into a mixture of C- and Oallylation adducts (129a : 129b ¼ 1 : 1). Both of them were useful because 129a underwent [3,3]-migration when the N-Boc group of the amide moiety was selectively removed by KOH. The terminal olen was then cleaved to give aldehyde, which was transformed into primary alcohol 130 and amine 131. The Pdcatalyzed Heck reaction of 132 with 2-methyl-3-buten-2-ol incorporated a prenyl group. Adding MsCl/Et3N triggered the establishment of the G ring. Finally, ()-communesin F was afforded aer the amide group of 133 was reduced and the A ring was formed.
Related prods:
CAS No. 100-06-1Ethanone, 1-(4-methoxyphenyl)- Catalog No.:AG000061 MDL No.:MFCD00008745 MF:C9H10O2 MW:150.1745 |
CAS No. 100-09-4Catalog No.:AG00005Z MDL No.:MFCD00002542 MF:C8H8O3 MW:152.1473 |
CAS No. 100-10-7Benzaldehyde, 4-(dimethylamino)- Catalog No.:AG00005Y MDL No.:MFCD00003381 MF:C9H11NO MW:149.1897 |
CAS No. 100-13-0Catalog No.:AG00005V MDL No.:MFCD00041254 MF:C8H7NO2 MW:149.1467 |
CAS No. 100-18-5Benzene, 1,4-bis(1-methylethyl)- Catalog No.:AG00005Q MDL No.:MFCD00008892 MF:C12H18 MW:162.2713 |
CAS No. 100-19-6Catalog No.:AG00005P MDL No.:MFCD00007355 MF:C8H7NO3 MW:165.1461 |
CAS No. 100-21-0Catalog No.:AG00005N MDL No.:MFCD00002558 MF:C8H6O4 MW:166.1308 |
CAS No. 100-26-5Catalog No.:AG00006P MDL No.:MFCD00006297 MF:C7H5NO4 MW:167.1189 |
CAS No. 100-27-6Catalog No.:AG00006O MDL No.:MFCD00010202 MF:C8H9NO3 MW:167.1620 |
CAS No. 100-31-2Benzoic acid, 4,4'-(1,2-ethenediyl)bis- Catalog No.:AG00006L MDL No.:MFCD00013994 MF:C16H12O4 MW:268.2641 |
CAS No. 100-32-3Catalog No.:AG00006K MDL No.:MFCD00003573 MF:C12H8N2O4S2 MW:308.3329 |
CAS No. 100-33-4Benzenecarboximidamide, 4,4'-[1,5-pentanediylbis(oxy)]bis- Catalog No.:AG00006J MDL No.:MFCD00599574 MF:C19H24N4O2 MW:340.4195 |
CAS No. 100-34-5Benzenediazonium, chloride (1:1) Catalog No.:AG00006I MDL No.: MF:C6H5ClN2 MW:140.5703 |
CAS No. 100-45-8Catalog No.:AG00006A MDL No.:MFCD00013778 MF:C7H9N MW:107.1531 |
CAS No. 100-48-1Catalog No.:AG000067 MDL No.:MFCD00006417 MF:C6H4N2 MW:104.1094 |
CAS No. 100-49-2Catalog No.:AG000066 MDL No.:MFCD00001510 MF:C7H14O MW:114.1855 |
CAS No. 100-51-6Catalog No.:AG000064 MDL No.:MFCD00004599 MF:C7H8O MW:108.1378 |
CAS No. 100-72-12H-Pyran-2-methanol, tetrahydro- Catalog No.:AG00006U MDL No.:MFCD00006624 MF:C6H12O2 MW:116.1583 |
CAS No. 100-75-4Catalog No.:AG00006R MDL No.: MF:C5H10N2O MW:114.1457 |
© 2019 Angene International Limited. All rights Reserved.