200,000+ products from a single source!
sales@angenechem.com


Home > Mass Spectrometry
Mass Spectrometry
A REVIEW ON THE DETERMINATION OF ISOTOPE RATIOS OF BORON WITH MASS SPECTROMETRY
Suresh Kumar Aggarwal1,2* and Chen-Feng You2,3
1
Fuel Chemistry Division, Bhabha Atomic Research Centre, Trombay,
Mumbai 400085, India
2
Department of Earth Sciences, National Cheng Kung University, Tainan,
Taiwan
3
Earth Dynamic System Research Centre, NCKU, Tainan, Taiwan
Received 20 October 2015; accepted 28 December 2015
Published online in Wiley Online Library (wileyonlinelibrary.com). DOI 10.1002/mas.21490
The present review discusses different mass spectrometric techniques—viz, thermal ionization mass spectrometry (TIMS), inductively coupled plasma mass spectrometry (ICPMS), and secondary ion mass spectrometry (SIMS)— used to determine 11B/10B isotope ratio, and concentration of boron required for various applications in earth sciences, marine geochemistry, nuclear technology, environmental, and agriculture sciences, etc. The details of the techniques-P- TIMS, which uses Cs2BO2þ, N-TIMS, which uses BO2—, and MC-ICPMS, which uses Bþ ions for bulk analysis or B— and Bþ ions for in situ micro-analysis with SIMS—are highlighted. The capabilities, advantages, limitations, and problems in each mass spectrometric technique are summa- rized. The results of international interlaboratory compari-son experiments conducted at different times are summarized. The certified isotopic reference materials avail- able for boron are also listed. Recent developments in laser ablation (LA) ICPMS and QQQ-ICPMS for solids analysis and MS/MS analysis, respectively, are included. The different aspects of sample preparation and analytical chemistry of boron are summarized. Finally, the future requirements of boron isotope ratios for future applications are also given. Presently, MC-ICPMS provides the best precision and accuracy (0.2–0.4‰) on isotope ratio measurements, whereas N-TIMS holds the potential to analyze smallest amount of boron, but has the issue of bias ( 2‰ to 4‰) which needs further investigations. # 2016 Wiley Period- icals, Inc. Mass Spec Rev 9999: 1–21, 2016.
Keywords: boron; mass spectrometry; thermal ionization mass spectrometry; inductively coupled plasma mass spectrometry; secondary ion mass spectrometry; laser ablation; isotopic reference materials; nuclear technology; marine science; geo- and earth sciences; B/Ca ratio
I.INTRODUCTION
Boron (B) is a light element that has two isotopes 10B and11B with 19.9 atom% and 80.1 atom% abundances, respectively.
Boron exists in solution in two forms—viz, trigonal boric acid B(OH)3 and tetrahedral borate anion B(OH)4—. These two forms equilibrate in solution, and their relative proportions depend upon the pH of the solution, as given below
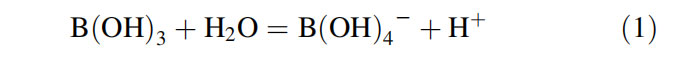
Trigonal B(OH)3 is predominant in acidic solutions whereas the tetrahedral anionic form is predominant in basic solutions. B(OH)3 is more enriched in 11B, whereas B(OH)4— is more enriched in 10B as given below. This is because of differences in the vibrational frequencies of the two boron isotopes and the molecular coordination between boron species in solution.
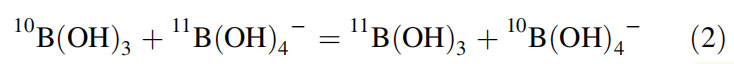
The anionic form gets incorporated into marine organisms (i.e., the corals or foraminifera shells) during their growth in the sea from calcium carbonates and thus 11B/10B isotope ratio in marine carbonates is a useful proxy for paleo-pH and thus paleo- CO2 (Vengosh et al., 1991; Hemming & Hanson, 1992). The dissociation constant (pKB) between these two forms of boron is
8.8 at 281.7 K and at 35.7 practical salinity unit. This dissocia- tion constant depends upon salinity, ionic strength, temperature, pressure (Dickson, 1990; Millero, 1995), and major ion chemis- try (Hain et al., 2015). Further, boron is geochemically incompatible and has high volatility, which leads to its migration and enrichment in Earth’s upper continental crust compared to primitive mantle (Tonarini, Pennisi, & Gonfiantini, 2009). A large isotope fractionation exists in solution with borate anion depleted in 11B. Experimentally, a value of 1.0272 0.0006 was obtained for boron isotopic equilibrium constant at 25˚C, salinity of 35 and ionic strength of 0.70 mol. kg—1 water (Klochko et al.,2006). Recently, an isotope fractionation factor of 26.0 1.0‰ at 25˚C was determined (Nir et al., 2015) for boron in sea water.
These data are corroborated by the ab initio molecular orbital calculations (1.0207–1.0360 by Oi (2000); 1.027 by Liu &
Tossell (2005); 1.020–1.050 by Zeebe (2005); 1.026–1.028 by Rustad et al. (2010)). The residence time of boron in sea water is about 14 Myr (Lemarchand et al., 2000) with d11B value of þ39.61 0.04‰ (Lemarchand et al., 2002a; Simon et al., 2006). Because the B residence time is much greater than the mixing time of the oceans (about 1,000 years), the isotopic composition of boron is uniform in sea water. It was also shown experimentally that sea water is isotopically homogeneous with respect to boron with a mean concentration of about 416 mmol. kg—1 (Foster, Pogge von Strandmann, & Rae, 2010). In view of the isotopic homogeneity, boron isotopes find increasing interest to understand environmental processes. A precision of better than 1‰ (2 SD) is needed in boron isotope ratio data for pH reconstruction with a resolution of about 0.1 pH (Foster et al., 2006). The high precision of 0.2–0.4‰ in boron isotope ratio data is required at low pH, because the slope of ~o11B curve gets shallower at low pH.
The isotope ratios of boron show large variations in nature (Fig. 1), and are of great interest to geo-, environmental, and marine scientists (Spivack, & Palmer, Edmond, 1987; Spivack, You, & Smith, 1993; Spivack & You, 1997; Lemarchand et al., 2002b; Kubota et al., 2015). These include studies in hydrology for ground water contamination by sea water (Barth, 1997); to assess the impact of artificial recharge on water sources; to identify mechanisms of adsorption/desorption on clay minerals; and in the environment as potential tracers of contamination of ground water with coal combustion residues, etc. (Davidson & Bassett, 1993; Vengosh et al., 1994; Ruhl et al., 2014; Warner et al., 2014). B isotope ratios are also used for provenance studies in archaeology (Devulder, Degryse, & Vanhaecke, 2013; Devulder et al., 2014, 2015) and hold potential in agriculture for crop plants (Wieser et al., 2001; Rosner et al., 2011; Geilert et al., 2015).
One of the isotopes of B (10B), has a high thermal neutron absorption cross-section (about 3,800 barns) and is, therefore, a useful nuclide in nuclear technology (IAEA Report, 1995; Subramanian, Suri, & Murthy, 2010). B2O3 dissolved in heavy water (D2O) is used as a liquid poison in the moderator system of pressurized heavy-water reactors (PHWRs) to control the reactivity. B4C enriched in 10B is used in control rods in nuclear reactors. Boric acid is also used in the primary cooling circuit of pressurized water reactors (PWRs), which require regular determination of isotopic composition and concentration of boron. Boron-alloyed steels are used as shielding materials for storage containers of irradiated nuclear fuel, reactor shielding, and in instruments used to detect neutrons. The nuclear reaction of 10B with thermal neutrons produces alpha particles.This property of B is exploited for boron neutron capture therapy (BNCT) to destroy cancerous cells in cancer treatment (Probst et al., 1997). In contrast, in other circumstances B may be less helpful, with the presence of B in aluminum, zirconium alloy, and stainless steel—clad materials or in nuclear fuel materials detrimental to neutron economy in nuclear reactors.
The utilization of B for palaeo-proxies, geology, archaeol- ogy, agriculture, nuclear technology, and medicine requires B isotopic composition data, which can be determined by various mass spectrometric techniques. The various mass spectrometric techniques used are thermal ionization mass spectrometry, inductively coupled plasma source mass spectrometry and secondary ion mass spectrometry. Because boron has a high first ionization potential (8.3 eV), generation of Bþ ions is not possible in TIMS. For the same reason, the yield of Bþ ions in an inductively coupled plasma (ICP) source is relatively low (about 50–60%), which necessitates solutions containing about 20 ppb of boron. Further, a difference of about 10% in the atomic masses of the two boron isotopes (i.e., 1 in 10) contributes to isotope fractionation or mass discrimination during their mass spectrometric analysis. In addition, the sticky nature of boron to various parts of the mass spectrometer causes memory effect or carry over in mass spectrometric analysis with ICPMS. This memory effect poses a severe problem when samples with widely varying boron isotopic ratios are analyzed, with an inductively coupled plasma source mass spectrometer.
The analytical chemistry of boron is challenging due to the propensity for contamination of boron from apparatus (e.g., borosilicate glass), chemicals, personnel, and laboratory envi- ronment during wet chemistry (Downing et al., 1998). At the same time, boron can be lost during separation and purification steps due to its volatile nature, which leads to incomplete recoveries. There can be fractionation between the two boron isotopes during chemical separations (Lemarchand et al., 2002a) and volatilization due to relative large mass difference between the two isotopes. Mass spectrometric techniques like P-TIMS and ICP-MS demand boron to be free from matrix and major abundant elements in the sample to achieve high sensitivity and high precision and accuracy to determine B isotope ratios. The small changes in 11B/10B isotope ratio are expressed in delta (d) notation (per mil), which is defined as
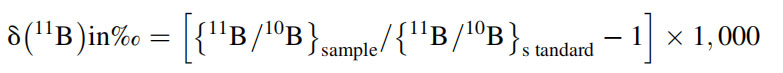

Precise and accurate determination of d11B in various samples (geological, biological, environmental etc.) demands the sample to be completely dissolved followed by separation and purification of boron. Great care has to taken to prevent B contamination even when working in an ultra-clean laboratory (Rosner, Romer, & Meixner, 2005). It was shown that boron- rich fibrous silicate glass filters should not be used in laborato- ries where samples with small amounts of boron are handled, and the glass filters should be replaced with synthetic poly- carbon filters made of polyethylene/polystyrole, which reduce the boron blank by an order of magnitude (Rosner, Romer, & Meixner, 2005). Also the laboratory ware to be used should be of Teflon, PFA, and properly leached prior to use (Downing et al., 1998). All chemicals should be of ultra-high purity, and acids used should be sub-boiling distilled.
In addition to great interest to obtain precise and accurate data on d11B, B/Ca ratio is important because this ratio in foraminifera samples is dependent on the partition coefficient of boron between sea water and calcium carbonate (calcite or aragonite). B/Ca ratio can, therefore, provide direct information on the [B(OH)4—/HCO3—] ratio of the sea water, because the amount of boron precipitated in the biological/inorganic carbon- ate is known to increase with increasing pH (Yu & Elderfield, 2007; Yu et al., 2010). Recent reports, however, show that B/Ca amount ratio is not a robust proxy and extensive careful studies are needed to evaluate its usefulness for the past ocean carbonate system (Allen & H€onisch, 2012; Babila et al., 2014; Henehan et al., 2015; Uchikawa et al., 2015).
The present review is written with the objective to summa- rize and highlight the scientific and technological advances that have taken place in the last two decades for the mass spectrometric analysis of boron (Hemming & Hanson, 1994; Al- Ammar, Reitznerova, & Barnes, 2000; Lemarchand et al., 2002a; Aggarwal et al., 2004; Albarede and Bear, 2004; Foster, 2008; Guerrot et al., 2010; Pennisi et al., 2011; Rae et al., 2011; McCulloch et al., 2014). The advancements are mainly because of two reasons: one is the availability of advanced fully automated multi-collector based high sensitivity thermal ioniza- tion and inductively coupled plasma mass spectrometers (MC- TIMS and MC-ICPMS) capable to provide high precision (better than 0.2‰) and secondly, an increasing interest in the bio-geochemistry of boron, particularly for marine carbonates, which serves as a proxy for paleo-pH and paleo-CO2 levels. Further, extensive experience in different international laborato- ries to determine isotope ratios of boron has highlighted the advantages and limitations of various methodologies to deter- mine accurate isotope ratios, that circumvent various problems like isotope fractionation in TIMS, memory effect in ICPMS, and validation of the methodologies with recently available certified isotopic reference materials. Also, techniques like laser ablation (LA) ICPMS and secondary ion mass spectrometry (SIMS) have added a new dimension to the in situ micro- analysis of boron in different geological and other solid materials. These aspects are also discussed in this review.
II.ANALYTICAL METHODOLOGY FOR EXTRACTION AND PURIFICATION OF BORON
Mass spectrometric analysis of boron with TIMS and ICPMS requires boron to be in a relatively pure chemical form to minimize matrix effects, organic isobaric interferences and suppression of ionization in the thermal ion source (Hemming & Hanson, 1994; Farhat, Ahmad, & Arafat, 2013). A number of approaches have been used to fulfil this requirement (Sah & Brown, 1997). The extraction and purification methods include solvent extraction, ion-exchange, micro-sublimation, etc. Among these methods, ion exchange procedure (Van Hoecke et al., 2014; He et al., 2015a) has been used more often. Micro- sublimation is also popular these days due to the advantages of efficient matrix removal, labor intensiveness, cost of consum- ables, procedural blank, and throughput (Van Hoecke et al., 2014). A brief description of these methods is given below.
The solid rock samples require extraction of boron that use fusion with NaOH, Na2CO3; pyro-hydrolysis, or acid dissolution (Aggarwal & Palmer, 1995). For example, boron can be extracted from borates and tourmaline (a complex alumina–silicate material) with fusion or pyro-hydrolysis. Pyro-hydrolysis consists of passing steam over the sample kept in a crucible (Pt) heated to high temperature (more than 1,000˚C) and collecting the condensate (Aggarwal & Palmer, 1995). This method provides a relatively pure fraction of boron unless the sample contains other volatile elements like sulfur, which require further purification. Fusion with Na2CO3 (flux: rock 8:1) or NaOH (5:1) offers the advantage that a large amount of solids that contain small amounts of boron can be taken up for fusion (Aggarwal & Palmer, 1995). Solid samples can also be dissolved with different acids like HF, perchloric acid, and nitric acid (Sah & Brown, 1997; Robinson, Skelly Frame, & Frame, 2014). Care has to be exercised with HF in view of the fact that BF3 is volatile in nature. Addition of small amounts of mannitol during acid dissolution prevents the loss of boron (Chen, Lin, & Yang, 1991; Nakamura et al., 1992). Care should be taken to use high purity acids, and also Teflon ware is a must with HF (Robinson, Skelly Frame, & Frame, 2014).
Boron extracted with the above procedures is further purified either with methyl borate distillation or more commonly with B- specific or non-specific ion-exchange resin columns (Aggarwal & Palmer, 1995). Distillation is based on the fact that trimethyl borate is volatile. The distillate will also include other volatile elements like halogens, Na, etc., which can be removed by passing the solution through a mixed cation-anion exchange resin bed treated with HCl and NaOH to convert the resin into Hþ and OH— forms, respectively. Boron can be eluted from the column with high purity water with cations and anions retained on the resin bed.
In biogenic carbonates, boron is generally separated and purified from the matrix with ion-exchange procedures. The foraminiferal carbonate tests (1–3 mg) are first cracked open with gentle crushing of the moist samples, in between the two clean glass plates (Barker, Greaves, & Elderfield, 2003; Rae et al., 2011; Henehan et al., 2013). The crushed samples are cleaned ultrasoni- cally with high purity water, methanol, high purity water (in sequence) to remove clay contamination. Oxidative cleaning is done for cultured and sediment samples, three to five times, with 1% H2O2 in 0.1 M NH4OH at 80˚C. Organic material is removed by oxidation with sodium hypochlorite (NaClO; 5%Cl), which is removed by repeated rinsing with high purity water. The cleaned samples are dissolved in dilute (0.1–0.5 M) HNO3 and Al/Ca ratio (<100 mmol mol—1) in the dissolved sample is monitored with high resolution ICPMS, to confirm effectiveness of the cleaning protocol. Al/Ca ratios of <50 mmol mol—1 were achieved with this protocol (Foster, 2008). Boron-specific anion exchange resin (Amberlite IRA 743) is conventionally used, either alone or preceded by a cation exchange resin column (Kiss, 1988; Leeman et al., 1991; Lemarchand et al., 2002a). Amberlite 743 contains a hydrophobic styrene backbone and a tertiary amine group (N- methyl glucamine), which is a weak base with a pKa value of about 7. The resin, therefore, behaves like an anion exchanger at pH <7 and absorbs only B(OH)4—. For example, separation of boron from seawater is given here (Foster, Pogge von Strandmann, & Rae, 2010). Seawater aliquots are mixed with a sodium acetate-acetic acid buffer to pH of about 5. The sample solution is loaded onto a micro-column (25 mL volume, 50 mesh resin). The matrix is washed with high-purity 18.2 MΩ MilliQ water, and B is eluted with 450 mL of 0.5 M HNO3. The B isotopic composition determined from different fractions collected with the IRA-743 ion exchange experiments showed that the first few fractions are enriched in 11B and the tail fractions are depleted in 11B (Lemarchand et al., 2002a). It is, therefore, important to achieve a high recovery to eliminate any isotope fractionation during ion-exchange purification. This is done by passing the sample slowly through the resin, which allows re-equilibration of the sample with resin in the column, after the initially absorbed borate is eluted slowly from the resin column. In addition, elution tails are checked for boron and the total procedural blanks are monitored for every column batch. Because B in the anionic form B(OH)4— is only absorbed onto the IRA 743 resin, some investigators propose that the B solution prior to loading on the resin column should have a pH of 10 (Rosner et al., 2011), which is achieved by adding alkali or ammonia, to eliminate isotope fractionation during boron purification. However, at this pH, Na, Mg, and Ca, if present in the sample, precipitate as hydroxide on the resin column and will elute along with boron (with HNO3 or HCl), and these ions have to be removed with another cation exchange step (Aggarwal & Palmer, 1995).
For foraminiferal shells, the amount of sample to be taken depends upon the species in view of different B concentrations in various foraminiferal species. For example, a few hundred mg (preferably 1–2 mg) of the benthic foraminiferal shells, with high B concentrations, are crushed, and rinsed and ultrasonicated with deionized water and methanol several times to remove clay materials (Yu et al., 2005). This step is generally followed by oxidative step to remove organic matter, in a clean laboratory. Oxidation with sodium hydroxide and hydrogen peroxide is done (Boyle & Keigwin, 1985; Rosenthal, Boyle, & Slowey, 1997; Barker, Greaves, & Elderfield, Yu & Elderfield, 2007; Foster et al., 2013; Holcomb et al., 2015). The cleaned shells are dissolved in 0.1 M HNO3 and the solution is taken up for isotopic analysis with MC-ICPMS. In another study, bleaching with sodium hypochlorite solution for 24 hr was done to oxidize the organic matter (Foster et al., 2006). These foraminiferal shells were again rinsed and ultrasonicated to remove NaClO and any dissolved salts. The cleaned foraminifer sample (calcite) was dissolved in 2 M HCl, by drop wise addition until complete carbonate dissolution (Foster et al., 2006). The purified solution was taken for N-TIMS analysis. In the case of coal-combustion residues (CCRs), effluents from leaching experiments on CCRs from a variety of coals were used to determine d11B (Davidson & Bassett, 1993; Williams & Hervig, 2004; Ruhl et al., 2014). The samples of surface water, effluents, and leachates were processed with a cation exchange resin (AG 50W-X8) to remove cations. The eluate was oxidized with H2O2 to remove organic matter and CNO complexes, prior to loading on the filament for N-TIMS analysis (Ruhl et al., 2014). Detailed studies are reported recently (Holcomb et al., 2015) for cleaning and pre-treatment procedures for biogenic and synthetic calcium carbonates formed in marine environments. Cleaning protocols that cause partial dissolution are troublesome for compositionally heterogeneous samples, and boron isotopes are always robust to sample pre-treatment cleaning procedures (Holcomb et al., 2015).
Studies were reported for evaporation and sublimation of boric acid to purify boron from organic-rich solutions (Gaillar- det et al., 2001). It was shown that slow evaporation of boron solution in dil. HCl medium, over a period of 10–12 hr, at 60– 65˚C gave complete B recovery without any isotope fraction- ation and boron loss. On heating the dried residue for more than 1 hr, loss of boron was observed. The recovery of boron in micro-sublimation experiments was confirmed with isotope dilution experiments, and the absence of any isotopic fraction- ation was confirmed with NIST certified B isotopic reference material (Gaillardet et al., 2001). It was also proved that mannitol addition is not necessary with this procedure of micro- sublimation. Boron loss was observed if the dried residue was heated beyond 1 hr, or a higher temperature was used for boron solution evaporation and sublimation. For these experiments with high temperature sublimation or heating of dried residue for longer durations, addition of mannitol was necessary to form a solid matrix of non-volatile boron ester to avoid the boron loss (Ishikawa & Nakamura, 1990). The micro-sublimation proce- dure was also found useful to separate boron from organic rich media, in contrast to other methods like the use of activated carbon, H2O2, organic matter specific resin or UV irradiation (Lemarchand et al., 2002a). Recently, this micro-sublimation was used (Wang et al., 2010; Liu et al., 2013; Pi et al., 2014) for rock samples, marine carbonate, sea water, and Porites coral samples with an aim to avoid introduction of organic residue to the purified sample through the ion-exchange procedure. The quantitative recovery of boron from sea water and coral sample (Liu et al., 2013) was confirmed by the standard addition method (Foster et al., 2006). The procedure involved the addition of a known amount of SRM 951a standard to a sample before micro- sublimation and to another set of the same sample after sublimation, followed by the measurement of 11B/10B isotopic ratios in the two mixtures to calculate the boron concentration. For the natural rock samples, doping with NaCl was necessary, prior to micro-sublimation, to recover boron from the digested silicates of the rock samples (Pi et al., 2014). The procedure is based on the fact that boron sublimates at a relatively low temperature (about 70˚C) to leave the organic and alkaline matrices in solution or solid form. Further separation with resins and additional H2O2 treatment is not required, and elimination of this separation step leads to low blanks in the separation procedure. The procedure involves taking a small volume of the boron-containing solution into the 5-mL conic bottom PFA vial, and setting it upside down so that the cap can be heated (about 70˚C for 12 hr) and the top conical end can be cooled for condensation of the evaporated boron. The benefits of low- blank, high-throughput, and quantitative recovery can be realized with the micro-sublimation method (Van Hoecke et al., 2014). A comparison of the micro-sublimation and anion exchange procedure for four different matrices, that is, Ca aqueous solution (20 g L—1), seawater, digests of spinach (100 g L—1) and silicate glass (10 g L—1), spiked with B standards showed good agreement in the chemical recoveries with the two methods, without any isotopic fractionation, except in spinach digests (Van Hoecke et al., 2014). Poor yield and isotopic fractionation observed in the spinach digests was attributed to the preferential adsorption of 10B, present as tetrahedral anion, to the organics present in undissolved portion of the sample.
For nuclear samples (e.g., titanium borides), a small amount of the sample is crushed and disintegrated with concentrated HNO3. The slurry is taken up in 0.5 M HNO3, and a small portion is loaded onto the filament and fused with rubidium carbonate on the filament, at alkaline pH, for boron isotope analysis with positive ions MC-TIMS (Rao et al., 2014). For the periodic analysis of heavy water from PHWRs to determine the concentration of boron, the solution is spiked with a known amount of pre-calibrated 10B enriched spike (Heumann, 1992) and pre-concentrated by heating under an infra-red lamp in a fumehood, prior to boron isotope analysis with P-TIMS. The solution is not allowed to dry-up, in the pre-concentration step, to eliminate the isotope fractionation effects. Solid boric acid or B2O3 samples for B isotope analysis are taken directly and fused with alkali carbonate on the filament. Solid samples like uranium oxide and aluminum, are first dissolved in suitable acids—for example, HNO3 for uranium and HCl for Al. Boron can be separated from different matrices in a 1 M HCl medium, by using solvent extraction with 0.65M solution of 2-ethyl- hexane 1,3 diol (EHD) in 10% chloroform (Rao & Aggarwal, 2008). In the nuclear samples (except in case of nuclear fuels and clad materials [aluminum, zircaloy, stainless steel], where boron is undesirable), the amount of boron present is generally not a limitation, but the blank and laboratory contamination need to be controlled and monitored carefully.
For crop plant samples, dry ashing at 600˚C in a microwave oven was used (Rosner et al., 2011). The ash was dissolved in 0.5 M HCl, and the solution was used to separate and purify boron with a 3-step ion chromatography procedure. The first step was to remove most of the cations with AG 50Wx8 resin on a polypropylene column. A boron solution was loaded under low- pH conditions so that boron was present as neutral B(OH)3 and is not adsorbed by the resin in contrast to all other cations. This step was proceeded by second and third steps of anion exchange purification with Amberlite IRA-743 boron-specific anion ex- change resin. In this step, the pH of the boron-containing solution was adjusted to >11 with aqueous NaOH so that B was present as B(OH)4— and was loaded onto the resin column. The boron fraction was eluted with 0.5 M HCl and mixed with mannitol (40 mg mannitol/1 mg of boron), followed by slow evaporation to dryness. No loss of boron was observed during dry ashing of these biological materials. A similar observation was reported (Noda & Ito, 2008) in coal combustion, where no loss of boron was observed under reducing conditions.
Among all the analytical methods discussed above, boron separation and purification with boron specific anion exchange resin IRA-743 is the most commonly used approach (He et al., 2015b) at various laboratories.
For foraminiferal shells, the amount of sample to be taken depends upon the species in view of different B concentrations in various foraminiferal species. For example, a few hundred mg (preferably 1–2 mg) of the benthic foraminiferal shells, with high B concentrations, are crushed, and rinsed and ultrasonicated with deionized water and methanol several times to remove clay materials (Yu et al., 2005). This step is generally followed by oxidative step to remove organic matter, in a clean laboratory. Oxidation with sodium hydroxide and hydrogen peroxide is done (Boyle & Keigwin, 1985; Rosenthal, Boyle, & Slowey, 1997; Barker, Greaves, & Elderfield, Yu & Elderfield, 2007; Foster et al., 2013; Holcomb et al., 2015). The cleaned shells are dissolved in 0.1 M HNO3 and the solution is taken up for isotopic analysis with MC-ICPMS. In another study, bleaching with sodium hypochlorite solution for 24 hr was done to oxidize the organic matter (Foster et al., 2006). These foraminiferal shells were again rinsed and ultrasonicated to remove NaClO and any dissolved salts. The cleaned foraminifer sample (calcite) was dissolved in 2 M HCl, by drop wise addition until complete carbonate dissolution (Foster et al., 2006). The purified solution was taken for N-TIMS analysis. In the case of coal-combustion residues (CCRs), effluents from leaching experiments on CCRs from a variety of coals were used to determine d11B (Davidson & Bassett, 1993; Williams & Hervig, 2004; Ruhl et al., 2014). The samples of surface water, effluents, and leachates were processed with a cation exchange resin (AG 50W-X8) to remove cations. The eluate was oxidized with H2O2 to remove organic matter and CNO complexes, prior to loading on the filament for N-TIMS analysis (Ruhl et al., 2014). Detailed studies are reported recently (Holcomb et al., 2015) for cleaning and pre-treatment procedures for biogenic and synthetic calcium carbonates formed in marine environments. Cleaning protocols that cause partial dissolution are troublesome for compositionally heterogeneous samples, and boron isotopes are always robust to sample pre-treatment cleaning procedures (Holcomb et al., 2015).
Studies were reported for evaporation and sublimation of boric acid to purify boron from organic-rich solutions (Gaillar- det et al., 2001). It was shown that slow evaporation of boron solution in dil. HCl medium, over a period of 10–12 hr, at 60– 65˚C gave complete B recovery without any isotope fraction- ation and boron loss. On heating the dried residue for more than 1 hr, loss of boron was observed. The recovery of boron in micro-sublimation experiments was confirmed with isotope dilution experiments, and the absence of any isotopic fraction- ation was confirmed with NIST certified B isotopic reference material (Gaillardet et al., 2001). It was also proved that mannitol addition is not necessary with this procedure of micro- sublimation. Boron loss was observed if the dried residue was heated beyond 1 hr, or a higher temperature was used for boron solution evaporation and sublimation. For these experiments with high temperature sublimation or heating of dried residue for longer durations, addition of mannitol was necessary to form a solid matrix of non-volatile boron ester to avoid the boron loss (Ishikawa & Nakamura, 1990). The micro-sublimation proce- dure was also found useful to separate boron from organic rich media, in contrast to other methods like the use of activated carbon, H2O2, organic matter specific resin or UV irradiation (Lemarchand et al., 2002a). Recently, this micro-sublimation was used (Wang et al., 2010; Liu et al., 2013; Pi et al., 2014) for rock samples, marine carbonate, sea water, and Porites coral samples with an aim to avoid introduction of organic residue to the purified sample through the ion-exchange procedure. The quantitative recovery of boron from sea water and coral sample (Liu et al., 2013) was confirmed by the standard addition method (Foster et al., 2006). The procedure involved the addition of a known amount of SRM 951a standard to a sample before micro- sublimation and to another set of the same sample after sublimation, followed by the measurement of 11B/10B isotopic ratios in the two mixtures to calculate the boron concentration. For the natural rock samples, doping with NaCl was necessary, prior to micro-sublimation, to recover boron from the digested silicates of the rock samples (Pi et al., 2014). The procedure is based on the fact that boron sublimates at a relatively low temperature (about 70˚C) to leave the organic and alkaline matrices in solution or solid form. Further separation with resins and additional H2O2 treatment is not required, and elimination of this separation step leads to low blanks in the separation procedure. The procedure involves taking a small volume of the boron-containing solution into the 5-mL conic bottom PFA vial, and setting it upside down so that the cap can be heated (about 70˚C for 12 hr) and the top conical end can be cooled for condensation of the evaporated boron. The benefits of low- blank, high-throughput, and quantitative recovery can be realized with the micro-sublimation method (Van Hoecke et al., 2014). A comparison of the micro-sublimation and anion exchange procedure for four different matrices, that is, Ca aqueous solution (20 g L—1), seawater, digests of spinach (100 g L—1) and silicate glass (10 g L—1), spiked with B standards showed good agreement in the chemical recoveries with the two methods, without any isotopic fractionation, except in spinach digests (Van Hoecke et al., 2014). Poor yield and isotopic fractionation observed in the spinach digests was attributed to the preferential adsorption of 10B, present as tetrahedral anion, to the organics present in undissolved portion of the sample.
For nuclear samples (e.g., titanium borides), a small amount of the sample is crushed and disintegrated with concentrated HNO3. The slurry is taken up in 0.5 M HNO3, and a small portion is loaded onto the filament and fused with rubidium carbonate on the filament, at alkaline pH, for boron isotope analysis with positive ions MC-TIMS (Rao et al., 2014). For the periodic analysis of heavy water from PHWRs to determine the concentration of boron, the solution is spiked with a known amount of pre-calibrated 10B enriched spike (Heumann, 1992) and pre-concentrated by heating under an infra-red lamp in a fumehood, prior to boron isotope analysis with P-TIMS. The solution is not allowed to dry-up, in the pre-concentration step, to eliminate the isotope fractionation effects. Solid boric acid or B2O3 samples for B isotope analysis are taken directly and fused with alkali carbonate on the filament. Solid samples like uranium oxide and aluminum, are first dissolved in suitable acids—for example, HNO3 for uranium and HCl for Al. Boron can be separated from different matrices in a 1 M HCl medium, by using solvent extraction with 0.65M solution of 2-ethyl- hexane 1,3 diol (EHD) in 10% chloroform (Rao & Aggarwal, 2008). In the nuclear samples (except in case of nuclear fuels and clad materials [aluminum, zircaloy, stainless steel], where boron is undesirable), the amount of boron present is generally not a limitation, but the blank and laboratory contamination need to be controlled and monitored carefully.
For crop plant samples, dry ashing at 600˚C in a microwave oven was used (Rosner et al., 2011). The ash was dissolved in 0.5 M HCl, and the solution was used to separate and purify boron with a 3-step ion chromatography procedure. The first step was to remove most of the cations with AG 50Wx8 resin on a polypropylene column. A boron solution was loaded under low- pH conditions so that boron was present as neutral B(OH)3 and is not adsorbed by the resin in contrast to all other cations. This step was proceeded by second and third steps of anion exchange purification with Amberlite IRA-743 boron-specific anion ex- change resin. In this step, the pH of the boron-containing solution was adjusted to >11 with aqueous NaOH so that B was present as B(OH)4— and was loaded onto the resin column. The boron fraction was eluted with 0.5 M HCl and mixed with mannitol (40 mg mannitol/1 mg of boron), followed by slow evaporation to dryness. No loss of boron was observed during dry ashing of these biological materials. A similar observation was reported (Noda & Ito, 2008) in coal combustion, where no loss of boron was observed under reducing conditions.
Among all the analytical methods discussed above, boron separation and purification with boron specific anion exchange resin IRA-743 is the most commonly used approach (He et al., 2015b) at various laboratories.
III.MASS SPECTROMETRIC TECHNIQUES USED TO ANALYZE BORON
A.Thermal Ionization Mass Spectrometry (TIMS)
1.Positive-TIMS
The high first- ionization potential of boron (about 8.3 eV) prevents the formation of Bþ ions with surface ionization in TIMS. Instead, alkali metal borate ions (M2BO2þ) are conven- tionally used for P-TIMS analysis of boron isotopes (DeBievre & Debus, 1969; Xiao, Beary, & Fassett, 1988; Vengosh et al., 1991; Nakano & Nakamura, 1998; Xiao & Wang, 1998; Ishikawa & Nagaishi, 2010). Different alkali metals (Li, Na, K, Rb, and Cs) have been used to generate positive meta-borate ions (Ramakumar et al., 1985; Spivack & Edmond, 1986; Nakamura et al., 1992; Ding & Zhao, 1994; Sahoo & Masuda, 1995; Catanzaro et al., 1970; Deyhle, 2001; Rao et al., 2008, 2010a). To efficiently generate M2BO2þ ions and reduce the intensity of alkali metal ions, a special loading procedure (e.g., loading of graphite as a promoter and an optimum B/alkali metal mole ratio) needs to be employed. Among the different alkali metals, Na and Cs metal borates are employed quite often in view of the fact that both Na and Cs are mono-isotopic and do not complicate the mass spectra. The 11B/10B isotope ratio can be conveniently calculated by measuring the ion intensities at 89/88 (Na2BO2þ) and 309/308 (Cs2BO2þ), with the exception to apply a small correction for an 17O contribution at masses 89 and 309. The correction of 17O contribution is straightforward and is accounted for by subtraction of 2 (17O/16O) isotope ratio value from the measured isotope ratio data. Between the two mono-isotopic alkali metals (Na and Cs), Cs is preferred in view of the high mass of the Cs2BO2þ ion, and this high mass minimizes the mass-dependent isotope fractionation, which is a source of variable systematic error in TIMS. This isotope fractionation is not a constant factor in TIMS, and depends on various parameters including the mass to charge ratio of the ions monitored in P-TIMS and N-TIMS. A high purity tantalum single-filament assembly is generally used for P-TIMS analysis. Because the use of Cs2BO2þ in a static mode of data collection was not possible with early models of TIMS, some laboratories continue to use Na2BO2þ ion in P-TIMS analysis. The continued use of Na2BO2þ ion is also partly due to the long experience of these laboratories in successful use of Na2BO2þ for isotope ratio measurements of boron (Rao et al., 2008, 2009; Rao, Parab, & Aggarwal, 2012).
For many years, the mechanism of formation of alkali metal borate ions remained elusive. During the past decade, a few interesting studies are reported by a few researchers (Lakshmi Narasimhan et al., 2013; Wei et al., 2011) that have shed light onto the mechanism of formation of M2BO2þ ions in TIMS. Studies were reported that used Raman spectroscopy and transmission electron spectroscopy (TEM) to compare the role of different carbon-based materials (e.g., graphite, carbon, fullerene, single-walled carbon nanotube SWNT; Wei et al., 2011). Based on the micro-structure properties of these four carbon-based materials, a surface-induced collision mechanism was proposed, and graphite gives the highest yield of polyatomic ions due to its perfect parallel and equidistant layers structure (only G-band observed with the lowest FWHM in Raman spectrum) and fullerene led to the lowest yield due to a block of their pathways (Wei et al., 2011). Studies performed on the reactions of B, B2O3, and B4C with Na2CO3 with transpiration thermogravimetry (TG), TIMS, and Knudsen effusion mass spectrometry (KEMS) showed that the formation of metaborate ion is a two-step phenomenon (Lakshmi Narasimhan et al., 2013). This two step process was concluded from the NaBO2 residue found in the TG experiments of all the three forms of boron (i.e., B, B2O3 and B4C with Na2CO3) at a temperature much lower than that for Na2CO3 alone, and also the high intensity of Na2BO2+ ion observed in KEMS.
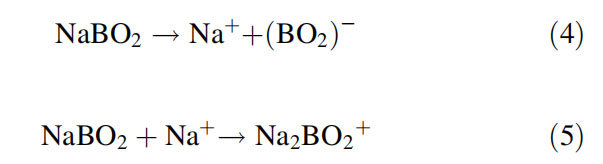
For TIMS analysis with Na2BO2+, it is generally accepted that the B/Na mole ratio needs to be strictly controlled on the filament within a narrow range when mixing with Na2CO3 or NaOH (Rao et al., 2008, 2009; Rao, Parab, & Aggarwal, 2012) to get good yield of Na2BO2þ ions. Studies (Rao et al., 2008) show that NaCl can also be used instead of Na2CO3 or NaOH, and in this case, the B/Na mole ratio need not be strictly controlled and could vary from 1 to 0.1. However, addition of mannitol, which forms an anionic complex with boron (70 mg of mannitol for about 1.7 mg of B), was essential, apart from loading of graphite on the filament. This relaxation in the strict control of the B/Na mole ratio is an advantage to analyze samples with unknown amounts of boron; e.g., in the case of ground-water samples. A subsequent study (Rao et al., 2009) demonstrated the robustness of a boron-mannitol complex on graphite-coated tantalum filament with Na2CO3 for solid samples and NaCl for solutions. With isotope dilution, P-TIMS was used to determine B at sub-ppm levels in uranium oxide samples (Rao et al., 2010b). For alloy samples of titanium boride, it was observed that graphite should be added onto the
filament only after fusion of the slurry of the alloy with Rb2CO3 (Rao et al., 2014) to get good intensity of Rb2BO2þ ions. Further, the possibility to perform internal normalization based on Rb isotope ratios was also demonstrated successfully (Rao et al., 2010a).
Isobaric interference limitation during TIMS analysis of boron with Cs2BO2þ is discussed in many papers (Hemming & Hanson, 1992; Aggarwal & Palmer, 1995). This isobaric interference is commonly encountered in organic samples—for example, marine carbonates, and lowers the d11B values. One of the ways to minimize this organic-matter (Cs2CNOþ) interfer- ence is to digest the sample with H2O2 (Grottoli et al., 2005) prior to loading on the filament. An experimental study (Wu et al., 2012) for Archaeo-cyatha fossil carbonates determined the exact nature of the organic matter with FT-IR, Raman, and TG-DSC-MS techniques. The results showed the presence of an acylamino (–CO–NH2) group in the carbonate samples, which would negatively bias the 11B/10B isotope ratios and lower the d11B values. It was also noted that, on external addition of acetamide (CH3CONH2), the B isotope ratio gradually reaches the accurate value on heating the filament to 1.8 A in TIMS, when the organic compound is consumed on the heated filament.
Another way to circumvent the isobaric interference (Wei et al., 2004) is to add 1% H3PO4 on the filament. The addition of H3PO4 acts as an ion depressor and prevents the formation of Cs2CNOþ in the presence of nitrate, probably by changing the morphology of boron compound. The temperature of TIMS analysis of B is also higher than that in the absence of ion depressor.
2.Negative-TIMS
Among different mass spectrometric techniques available, N-TIMS has the highest sensitivity for isotopic analysis of boron, and can conveniently analyze samples that contain one ng or smaller amounts (150 pg) of boron (Duchateau & DeBievre, 1983; Zeininger & Heumann, 1983; Heumann & Zeininger 1985; Vengosh, Chivas, & McCulloch, 1989; Hem- ming & Hanson, 1992, 1994; Heumann et al., 1995; Sonoda et al., 2002; Shen & You, 2003; You, 2004). Samples like foraminifera with 1–2 ng of boron can be easily analyzed with this method. During N-TIMS, a single-filament assembly made of a zone-refined high-purity rhenium filament is used to load the sample solution. All efforts are made to minimize the isobaric interference of CNO— at m/z 42, which corresponds to 10BO2—. This minimization of isobaric interference is neces- sary, because even 0.1% of CNO— in the ion beam will lower the 11B/10B isotope ratio by 4.7‰ compared to the exact value (Tonarini, Pennisi, & Gonfiantini, 2009). Samples are treated with high-purity hydrogen peroxide to remove CNO complexes and any organic matter (Pelejero et al., 2005; Foster et al., 2006). In addition, signal is monitored at m/z 26 (CN—) using secondary electron multiplier (due to its higher sensitivity compared to a Faraday cup) to confirm the absence of any signal at m/z 26 and thus of no interference at CNO—. All operations should be done in a laminar-flow clean hood equipped with boron-free PTFE HEPA filters to minimize boron contamination of the sample (Rosner, Romer, & Meix- ner, 2005). For N-TIMS, boron-free sea water is generally loaded on the filament to enhance ionization efficiency, because the electronic work function of the filament is lowered by salts present in sea water. The boron-free sea water is prepared with Amberlite IRA 743 ion-exchange resin (Barth, 1997), which introduces some organics from the resin. Another method with an alternative loading solution prepared by combining high- purity single element standard solutions of Ca2þ, Mg2þ, Naþ, and Kþ in 5% HCl with proportions similar to those present in sea water was found to give encouraging results. This loading solution eliminates the risk of isobaric interference from CNO— and provides the possibility to perform automatic data collec- tion (Dwyer & Vengosh, 2008).
Owing to the low mass of BO2— ions, there is a problem of isotope fractionation in TIMS. This isotope fractionation is a serious problem in B because B has only two isotopes. Different methodologies are adopted to circumvent this drawback. The 11B/10B isotope ratio is determined with any of the three approaches during TIMS data acquisition. These approaches are(i) follow a strict protocol developed previously with certified isotopic reference material like NIST-SRM-951; (ii) use total evaporation (TE) (Foster et al., 2006); and (iii) by internal normalization (IN) based on 18O/16O ratios determined in-situ from ReO4— ions, from each filament loading, prior to mass spectrometry for boron (Aggarwal et al., 2009b). The total evaporation approach to take care of isotope fractionation in TIMS involves integration of the ion current until complete exhaustion of the sample on the filament and was proposed (Kanno, 1971) for elements with high ionization efficiencies and follow Rayleigh distillation as in the case of single-filament assembly. This total evaporation method requires less than 1 ng of boron to be loaded on the filament due to the very high sensitivity of N-TIMS of boron, and completion of the analysis within a reasonable time. This loading of about 1 ng of boron demands a strict control of boron blank during all stages of sample handling. Internal normalization is based on the invariant nature of one of the isotope amount ratios (e.g., 18O/16O) present in the sample on a given filament. The isotope fractionation factor is calculated from the 18O/16O isotope ratio by using the 187Re/185Re isotope ratio determined from ReO4— ions from the same filament. With the 18O/16O isotope ratio from the ReO4— ion, the fractionation factor is calculated for the 18O/16O ratio observed with the BO2— ion, and this factor is applied to account for isotope fractionation in the boron isotope ratio. The application of this internal normalization methodology was demonstrated on NIST-SRM-951 isotopic reference material, in sea water as well as in coral samples (Aggarwal et al., 2009b).
It has been reported that determination of d11B in forami- nifera samples with TE-NTIMS gives a bias with respect to the data obtained with MC-ICPMS. This bias is attributed to the incomplete decomposition of the organics that arise from foraminifera tests. This inference is supported by the observa- tion that the same solution during repeated analysis with TE- NTIMS over a period of a few months gives a d11B value lower by 6‰ probably due to the hydrolysis of organic matter by the acid present in the solution (Foster, 2008). In the studies performed previously with N-TIMS (H€onisch & Hemming, 2005; Pelejero et al., 2005; Hemming & H€onisch, 2007), each sample was analyzed with several independent filament loadings (at least three times), with strict protocol of filament heating and data acquisition, to confirm the absence of analytical artifacts such as excessive isotopic fractionation and isobaric interfer- ences on mass 42 by the organic matter. The mass spectrometric analysis runs which showed fractionation and/or isobaric interference were discarded, until three acceptable runs data showing no fractionation (less than 0.1‰) in 20 min of acquisition were obtained and averaged. In the light of these discussions, further studies are necessary to make the N-TIMS method robust and rugged to achieve precision and accuracy of better than 0.2–0.4‰, to be useful for paleo-proxy studies.
B.Inductively Coupled Plasma Mass Spectrometry (ICPMS)
ICPMS is a useful and quite convenient mass spectrometric method to determine the isotope ratios and concentrations of different elements in a variety of matrices. Solutions are analyzed with a pneumatic nebulizer (PN), whereas solids can directly be analyzed with laser ablation (LA). A number of commercial vendors—(e.g., Agilent [Japan], GBC Scientific [Australia], Nu Instruments [UK], Perkin Elmer [USA], Thermo Fisher [Germany]) supply a variety of ICPMS systems with different kinds of mass analyzers. These ICPMS instruments include quadrupole-based ICPMS with or without a collision or reaction cell, time-of-flight based ICPMS, high resolution double-focusing electrostatic and magnetic analyzers based ICPMS (Element 2 from Thermo and AttoM from Nu Instru- ments), and multiple collector magnetic sector based ICPMS (NuPlasma and Neptune).
Among the various types of ICPMS systems, Q-ICPMS and ToF ICPMS are employed to determine concentrations of different elements, and their isotope ratios in environmental and biological applications that do not demand high precision. These quadrupole- and ToF-based ICPMS systems are more cost- effective compared to MC-TIMS and MC-ICPMS, but provide lower precision and poor accuracy (Gregoire, 1987; Lu, 2014) in isotope ratio measurements due to Gaussian peak shapes. Therefore, these Q-ICPMS and ToF-ICPMS are not employed to determine precise and accurate isotope ratios, but are used mostly for multi-elemental analysis to determine the trace and ultra-trace concentrations of different elements in various matrices. Most of the quadrupole-based ICPMS instruments are equipped with either a dynamic reaction cell (DRC) or a collision cell to overcome the isobaric interferences from the atomic isobars as well as to eliminate polyatomic interferences (Tanner & Baranov, 1999). This removal of isobaric interference is based on the preferential chemical reaction of the analyte ion or the interfering ion with a reactive reagent gas (e.g., oxygen, ammonia, etc.), which shifts the m/z ratio and thereby chemi- cally resolves isobars. In addition, polyatomic molecular interfering ions are dissociated due to collisions with the reagent gas. Another approach is with kinetic energy discrimination with an inert gas. These systems provide an abundance sensitivity (contribution of high-abundant peak of mass M to adjacent peak at mass M 1) of 107.
A novel QQQ-ICPMS (from Agilent, 8800) with an octo- pole reaction cell is introduced recently in the market (Agilent, 2013). This triple-quad ICPMS eliminates the atomic and polyatomic isobaric interferences with the use of a collision or reaction gas in the second quadrupole, and in addition, gives a high abundance sensitivity of more than 108. This instrument needs to be evaluated for its capability, in terms of precision and accuracy, to determine boron isotope ratios.
Single-collector magnetic sector based HR-ICPMS is quite popular to analyze samples for determination of concentrations of various trace constituents, because the use of double focusing electrostatic and magnetic analyzers allows to eliminate most of the polyatomic isobaric interferences commonly encountered at m/z below 80 in Q-ICPMS. MC-ICPMS systems from Thermo as well as Nu Instruments are available at different international laboratories, and are the best to perform isotopic analysis of various elements with the highest precision (0.01% or better) and accuracies. HR-ICPMS and MC-ICPMS both have the options to use these systems at low, medium, and high mass resolutions. Experience in different international laboratories over the past decade has demonstrated that samples with a lower chemical purity, compared to that required for MC-TIMS analysis, can be analyzed conveniently with MC-ICPMS to make the latter a more popular analytical tool compared to MC- TIMS (Yang, 2009). The MC-ICPMS-based techniques are also characterized by their fast analyses that lead to high throughput, subjected to optimization of different operating parameters of ICPMS and matching the matrices of sample and standard. Boron is analyzed as Bþ ions, taking care of a tail contribution from 12Cþ, memory or carry-over effect, and control of the laboratory and procedural blank values. The standard sample bracketing (SSB) approach is popularly used to take care of mass discrimination and signal drift during data collection. Among the various types of ICPMS instruments available commercially, only the MC-ICPMS allows the determination of 11B/10B isotopic ratios with relative precision values of 0.2–0.4‰ required in paleo-proxy studies and is, therefore, the most popular with bio- and geo-scientists (Rehkamper, Sch€onbachler, & Stirling, 2003). However, to achieve such high precision in the isotopic ratio data, the non-spectroscopic matrix effects, for example, the effect of Na contamination and other matrix elements present in the sample have to be checked. Because B is a light element, and there is a difference of about 10% in the atomic masses of the two isotopes, the presence of Na (Na/B molar ratio of 20,000 and Na concentration of 6,000 ppm in solution with 150 ppb of boron) was reported to give 12% suppression in the B count rate and 8% increase in the observed 11B/10B isotopic ratio (Gregoire, 1987). Additionally, a compari- son of the matrix effects with Na, Cs, and Pb showed that the presence of heavy element causes the highest suppression effect to the intensity of boron (Gregoire, 1987). Therefore, separation and purification of boron, from bulk of matrix elements or organic impurities is one of the important pre-requisites to the determination of boron isotopic ratios with high precision and accuracy.
C.Mass-Bias Effect
The mass-bias effect (also known as isotope fractionation or mass discrimination) occurs in ICPMS due to space-charge effects or in the high vacuum region between sampler and skimmer cones. This effect leads to preferential extraction and transmission of the heavier isotope compared to the lighter one and, therefore, the isotope ratio data are positively biased with respect to the heavier isotope (Albarede et al., 2015). In case of boron, a mass-bias effect of 4–9% per atomic mass unit was reported with an MC-ICPMS (Thermo Axiom) system (Aggar- wal et al., 2003). However, this mass discrimination is quite stable, and can be easily corrected for with the SSB approach (Foster, 2008). Sample solutions with 20–50 ppb of boron and NIST SRM-951 boron standard of similar concentration are analyzed. A typical sequence involves the analysis of blank, standard, sample, standard, blank. This is repeated two times for every sample and the average d-11B value is calculated. It is important that the sample and the standard are matrix-matched because the presence of large amounts of other elements (e.g., Na) and the acid strength used to introduce the sample will give rise to matrix effects affecting the B isotope mass fractionation
D.Memory Problem
As stated previously, determination of B isotope ratios or B concentration is plagued by the carry-over effect or memory problem due to the adhering tendency of B to the instrumental components. This memory effect has been noticed by several researchers and a number of approaches have been used to minimize this effect. These approaches include a wash between analyses with ammonia solution, inject NH3 gas into the spray chamber during analysis (Al-Ammar, Gupta, & Barnes, 1999, 2000), wash with NaF, mannitol (C6H8(OH)6), a wash with
H2O2 (10%), HF (0.3%), HNO3 (0.65 N), and finally HNO (0.05 N; Lecuyer et al., 2002). Nebulized ammonia reacts with boric acid to form ammonium borate, which is non-volatile, and is, therefore, washed out of the spray chamber without the production of Bþ ions. Detailed studies were performed on the introduction of ammonia gas into the spray chamber to optimize the NH3 flow rate (Al-Ammar, Gupta, & Barnes, 2000). It was shown that a wash time of 2 min is sufficient to get rid of the boron memory. At the same time, an enhancement in the boron signal intensity was observed, which was attributed to an increase in the thermal conductivity due to hydrogen formation from ammonia or charge exchange reaction between the positively charged nitrogen species and boron atoms (Al- Ammar, Gupta, & Barnes, 2000). Also, no analyte loss, matrix precipitation, or nebulizer blockage was observed. Most MC-ICPMS laboratories use dilute HNO3 (0.5 M) as the wash solution, for 2 min, in between the samples to minimize the memory effect in B isotope ratio measurements.
Several other approaches were tried in the past to circum- vent the ubiquitous memory problem in B concentration determinations in various matrices. These methods cannot be used in B-isotope ratio measurements with MC-ICPMS, because of the matrix effects induced by the introduction of various chemicals. A rinse solution of 0.25% (w/v) mannitol in 0.1 M NH3 was used to eliminate the boron memory effect after a 5- min wash (Sun et al., 2000) to determine B in biological samples (e.g., serum, plasma, and urine). Mannitol has been used by several researchers in view of the tendency of boron to form complexes with alcohols and polysaccharides. A rinse solution of ammonia, EDTA, surfactant Triton X100, and H2O2 was used (Wright et al., 2008) for standard sample introduction system during analysis of boron with Q-ICPMS in soil/plant samples. A mixture of 0.1 M HNO3 and 0.3M HF was used with a Teflon spray chamber (Misra et al., 2014). A demountable direct- injection high-efficiency nebulizer (d-DIHEN; Bellato, Mene- gario, & Gine, 2003) was also used to circumvent the memory effect. A comparison on the Meinhard pneumatic nebulizer with a Scott-type double-spray pass chamber and direct injection nebuliser (Smith et al., 1991) showed the advantages of DIN from the point of view of low sample consumption as well as quick wash with negligible memory. In the case of a pneumatic nebulizer, a signal of 2% of initial boron intensity, which decreased very slowly, was seen even after washing for eight minutes with a 2% HNO3 solution. With DIN, because liquid samples are transported inside a capillary by pumping at high pressure and by introducing its aerosol directly into the plasma, no spray chamber is required.
The best way to confirm the complete elimination of any memory effect is to make multiple injections of natural boron solution or NIST standard SRM-951, enriched 10B (d11B value of at least 100 or more) solution, and again natural B solution with similar concentrations of boron (Aggarwal et al., 2015, unpublished results).
In addition to extended washing with 2–3% HNO3, it was suggested that an on-peak zero blank correction (Wei et al., 2014) should be applied to account for the memory effect, particularly when analyzing samples with widely varying d11B values as well as for samples with extremely small amounts (ng or less) of boron. The corrected 11B/10B isotope ratio of the sample is calculated as:
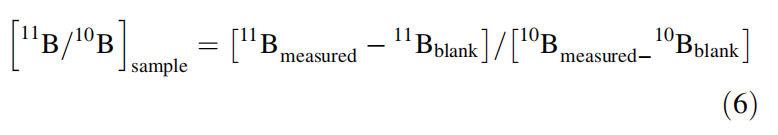
E.Laser Ablation ICPMS (LA-ICPMS)
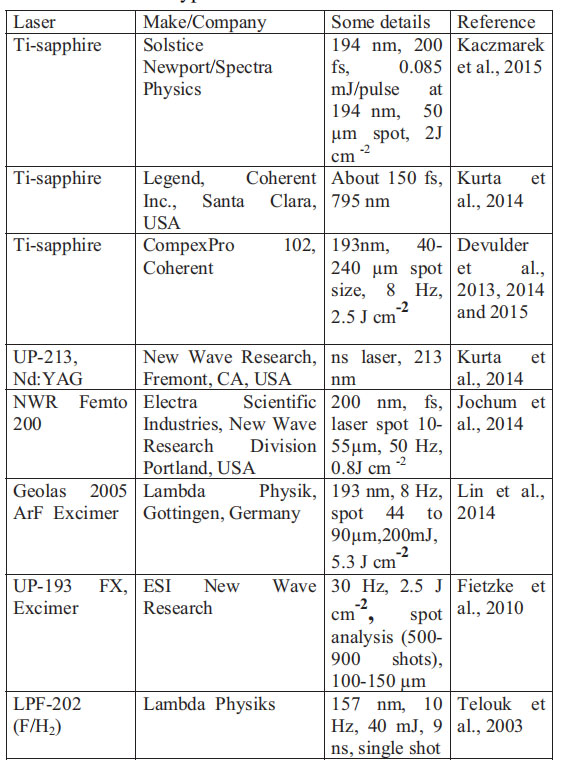
Laser ablation ICPMS depends on the use of a laser to generate an aerosol of the material in an ablation cell. The generated vapors are transported with He, Ar, or Ar N2 gas into the argon plasma of ICP, where they are atomized and ionized. Different kinds of lasers have been used (Table 1), and involved a 1,024 nm solid state Nd-YAG laser to start with, 532 nm (second harmonic), 266 nm (third harmonic), 213 nm (mixing of third
TABLE 1. Different types of lasers used in LA-ICPMS for boron
ANGENE is pledged to providing quality specialty chemicals for use in research and development and commercial manufacturing:
© 2019 Angene International Limited. All rights Reserved.